Introduction to Evolutionary processesThe microbiome is the aggregation of all the microorganisms in a system; this can be the human gut microbiome or a biofilm on a rock in a stream. Regardless of the system, the microbes inhabiting that environment are constantly competing for resources (e.g., physical space, nutrients, etc.). Thus, community assembly can be highly deterministic if the environmental conditions and interspecific interactions determine which species persist in a local community. Typically, we associate microbe's traits to the environment, as these traits underlie an organism's response to both biotic and abiotic factors. These traits are inherently based on evolutionary history. So a major question is how can we observe evolution in these systems? For one, environmental disturbances can lead to community turnover - this is typically associated with ecological processes. However, is it time to start reevaluating these processes from an evolutionary standpoint? After all, a change in allele frequency is the definition of evolution. Typically, evolution is studied at the population level, but this is increasingly hard to do in microbiomes. What defines a natural population? Moreover, with rapid population sizes and fast generation times, how can we disseminate ecological and evolutionary processes in microbiomes? Maybe they are one in the same...This month's reading list will highlight some recent (and not so recent) papers addressing evolutionary processes and how they relate to microbiomes. I will not go into population-level processes since I have covered them in other posts here and here. Instead, I want to focus on the challenges in microbiome studies on the aggregate. Human Microbiome
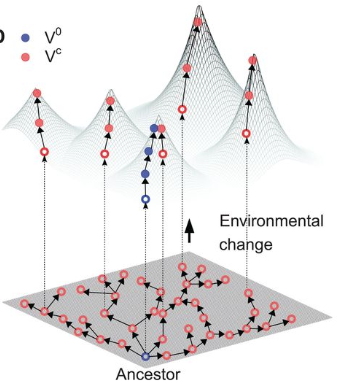
Niche expansion and adaptationIf mutations can lead to increased fitness, what prevents the "optimal" phenotype from dominating a given system. Certainly, a generalist approach can utilize diverse resources and outcompete specialists with limited resources and narrower niche breadth. The next paper address the idea of evolvability [2] with respect to the generalist v. specialist strategy. In particular, the cost of a generalist resides in their reduced capacity to evolve to changing environments. In other words, selection is weakened or relaxed in the generalist strategy since it can readily access a wider range of niches, albeit at local peaks of lower fitness. The authors describe this in the context of a fitness landscape where specialists can rapidly respond to environmental changes whereas the generalist must maneuver through a rugged fitness landscape resulting in a "lag load" to peak fitness.
Observing evolution and molecular adaptationAll of this has been amazing work and definitely worth pursuing more in microbial systems. But a major question is how to observe these evolutionary dynamics in high resolution. This last paper utilizes a barcoding system to track adaptation in yeast [5]. Using this method, the authors can re-barcode each population to tract specific lineages arising from mutations. Initially the ancestral population accumulates a distribution of beneficial mutations. Lower fitness clones die out and are replaced by newer, other beneficial mutants that maintain diversity in the population. This distribution of continuously generated variation provides a broad distribution across a fitness mean. Papers:1. Garud NR, Pollard KS. (2019). Population genetics in the human microbiome. Trends in Genetics
2. Bono LM, Draghi JA, Turner PE. (2019). Evolvability costs of niche expansion. Trends in Genetics 3. Venkataram S, Monasky R, Sikaroodi SH, Kryazhimskiy S, Kacar B. (2019). Evolutionary stalling in the optimization of the translation machinery. bioRxiv 4. Zheng J, Payne JL, Wagner A. (2019). Cryptic genetic variation accelerates evolution by opening access to diverse adaptive peaks. Science 365: 347-353. 5. Nguyen Ba AN, Cvijovic I, Rojas Echenique JI, Lawrence KR, Rego-Costa A, Liu X, Levy SF, Desai MM. (2019). High-resolution lineage tracking reveals travelling wave of adaptation in laboratory yeast. Nature 575: 494–499.
0 Comments
IntroductionI haven't posted in a while because I have been trying to find new and exciting papers a little out of my comfort zone. And it just so happened that Scripps hosted National Academy member, Diane Newman, this week for a special seminar sponsored by the graduate students in the Biotechnology Training Fellowship. Almost all of my interest has been driven by a graduate student in my current lab, Doug Sweeney, who is actively looking at the mechanisms of electron transfer in marine sediment bacterium. If you are interested in any of these topics, definitely feel free to email him!
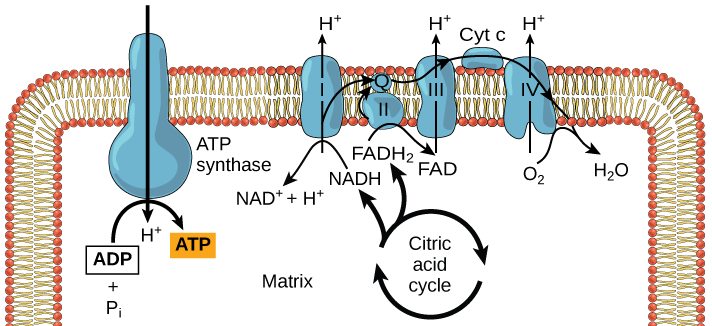
Redox potential are everything in microbial communitiesBefore I jump into the chemistry side of things, I first wanted to give a brief overview of why electron transfer is so imperative. Most of you probably know this, but this process of transferring electrons enables cells to maintain cellular function and generate energy for additional processes. But since I am not a chemist, I obviously want to take a step back very briefly to highlight how important these processes can be at multiple scales, especially when we consider entire communities! A recent paper by Ramirez-Flandes et al. (2019) assessed metagenomes from 18 biomes to determine what distinguishing characteristics delineate microbial communities [1]. Most of us are aware that taxonomic signatures can illuminate certain microbial biomes (e.g., Cyanobacteria are usually associated with marine samples). However, this paper decided to turn to functional characterization to differentiate various microbial biomes. What they found was that oxidoreductase gene profiles separated the biomes into three main clusters: anoxic biomes (e.g., host-associated and marine sediments), aquatic biomes, and soil-associated biomes. Within the oxidoreductase gene profiles, different biomes were associated with various groups of oxidoreductases related to substrate availability and stress conditions. Redox processes alone delineate microbial communities! Why are redox processes so important?I started with this first paper because it nicely outline that various biomes present various challenges for the bacteria that reside there. For example, in the absence of oxygen, how do organisms maintain their cellular processes? One way is fermentation, but this leads to toxic byproducts and is not a sustainable option. Another is the use of alternative electron acceptors. In soils, sediments, host-associated, microbes are constantly dealing with anoxic conditions and yet we find traditionally described aerobic organisms. Some organisms have adapted to utilize redox-active minerals that are abundant in certain biomes, like iron or manganese in soils. This is absolutely fascinating, microbes have evolved novel strategies to exchange electrons with minerals by transferring electrons from the cytosol to the exterior of the cell [for more see this review [2]]. 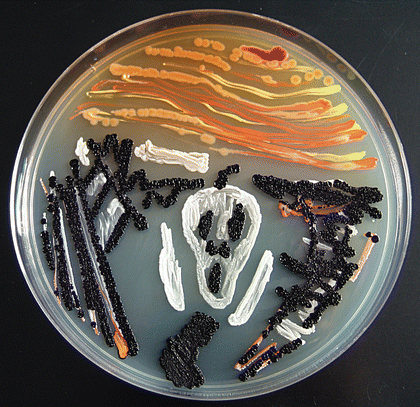 So how do these microbes transfer electrons outside the cell? The above image [3] depicts the beautiful pigments bacteria are capable of producing on a plate that most of us take for granted. But what is the role of these pigments? Some may be related to UV or desiccation, but others often change colors which is indicative of a redox reaction. This led to the topic of the entire post, the use of these molecules as extracellular electron shuttles. Some known shuttles are the natural products, quinones, phenazines, and flavins, which shuttle electrons to an acceptor. Glasser et al. [3] found that phenazines in particular are widespread along with the transcription factor, SoxR, that senses redox-active metabolites. From Dr. Newman's seminar this week, she discussed (in fascinating detail) how these phenazines in biofilms interact with eDNA (or dead cell DNA) to stabilize redox reactions and allow for the anaerobic survival of the producing organisms to maintain cellular viability. These discoveries really revolutionize our understanding of microbial metabolism. But the biggest question I always had is the production of such natural products must be expensive, especially considering their role is essential in stressed conditions. And even if production is sustained, how can the cell ensure an adequate return on its investment; in other words, how can the extracellular molecule not be lost to the environment. Dr. Newman presented an amazing solution to this problem by addressing shuttle diffusion gradients, electron hopping, and more [see her review [3] and stay tuned for some forthcoming work from her lab]. Why do microbes go through all this trouble???Finally, this question brings me to the Journal Club paper of the week [4]. Why are microbes evolving these intricate solutions to a seemingly simple problem of where to deposit electrons. As a reminder, the entire point of transferring electrons is to generate ATP, but to do so requires the use of electron shuttles such as NADH to carry electrons from one reaction to another. This essential molecule then can be used as a reducing agent to donate electrons and become oxidized as NAD+, ready to accept more electrons and continue the cycle. In the absence of a terminal electron acceptor, such as oxygen, NADH will build up in the cell. 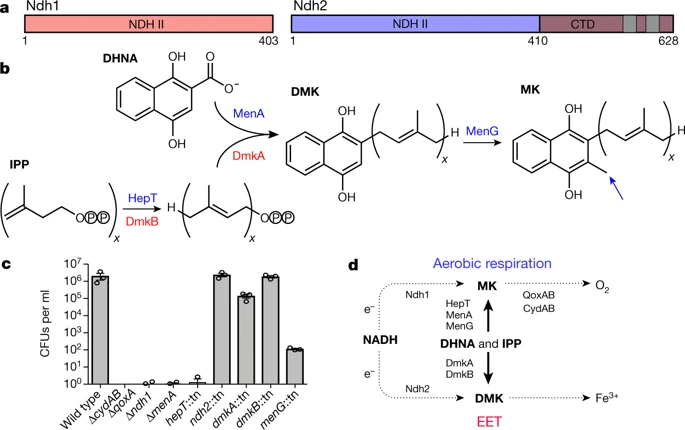 What would be a good strategy to deal with NADH buildup? Well in a paper by Light et al. [4] the authors identified a parallel electron transfer pathway in anaerobic conditions in the pathogen Listeria monocytogenes. In the presence of oxygen, normal aerobic respiration occurs with an essential enzyme for metabolism, NADH dehydrogenase (Ndh1 in above figure), catalyzing electron exchange to a quinone derivative, the first step in the electron transport chain. Under anaerobic conditions, this process cannot continue as NADH cannot be recycled. So, a parallel solution evolved using another NADH dehydrogenase (Ndh2 in above figure) enables NAD+ concentrations to be restored. Using transposon mutants the authors show cell viability using knockouts of these essential genes (panel C); for instance, in the presence of oxygen the knockout of Ndh2 (or any other genes in the anaerobic pathway) does not inhibit cellular growth. Under anaerobic conditions, the demethylated quinone derivative (DMK) accepts electrons from NADH via Ndh2. The authors very thoroughly outline the subsequent electron transferring system where DMK donates electrons to membrane proteins and finally to environmental flavins to shuttle electrons and finally to extracellular acceptors, like Fe(III). More and more, it appears that these innovative strategies to deal with electron transfer are a fundamental issue in all microbial life, not limited to specialized processes in mineral-respiring bacteria. All bacteria require cell viability and processes. These processes are driven entirely by redox potential which is mediated by electron shuttles. This really blows my mind as I have never considered the magnitude of the electron transport chain (and the various innovations) to deal with the recycling of electron shuttles. Papers:1. Ramirez-Flandes S, Gonzalez B, Ulloa O. (2019). Redox traits characterize the organization of global microbial communities. Proceedings to the National Academy of Sciences 116(9): 3630-3635.
2. Shi L, Dong H, Reguera G, Beyenal H, Lu A, Liu J, Yu HQ, Frederickson JK. (2016). Extracellular electron transfer mechanisms between microorganisms and minerals. Nature Reviews Microbiology 14: 651-662. 3. Glasser NR, Saunders SH, Newman DK. (2017). The colorful world of extracellular electron shuttles. Annual Review of Microbiology 71: 731-751. 4. Light SH, Su L, Rivera-Lugo R, Cornejo JA, Louie A, Iaverone AT, Ajo-Franklin CM, Portnoy DA. (2018). A flavin-based extracellular electron transfer mechanism in diverse Gram-positive bacteria. Nature 562: 140-144. Introduction to microbial biogeographyUtilizing a biogeographic framework is extremely powerful as biologists can understand the ecological and environmental factors driving the distribution of taxa. In addition, extensions of this include phylogeography where evolutionary biologists can understand barriers to dispersal and geological events that may contribute to species' distributions. More recently, these patterns have been observed for microbes. However, microbial biogeographic patterns are typically associated with environmental factors at the community level. This is in stark contrast to the traditional work in plants and animals, where species' distributions and/or populations are described. Previously, I wrote a commentary with my PhD advisor, Jennifer Martiny, going over some of these ideas [1]. Recently, I have seen a lot more papers moving beyond community-level patterns and trying to identify the ecological and evolutionary processes structuring the distribution of microbial taxa. So, for this month, I thought everyone could take a deep dive into some of these amazing papers. I tried to find papers referencing soil, marine, and host-associated bacteria to give a little bit of something for everyone. This list is in no way inclusive, so please feel free to share other papers you have seen as well! I will be more than happy to add to this list. Global-level patternsFirst couple of papers I want to highlight are looking at the global distribution of microbial taxa. To start, we can look at soil bacteria. A lot of papers have previously shown that, at global levels, various edaphic properties can structure soil communities, including pH, temperature, and salinity. Greenlon et al. examined the phylogenetic diversity of the nodule-forming genus, Mesorhizobium, by sampling chickpeas across various soil types, climates, continents, etc. [2].
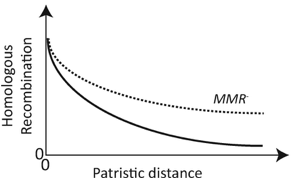
The second paper dives into maybe the most abundant organism on the planet, the SAR11 clade. This order has a vast global distribution in oceans with finer clades exhibiting differential geographic patterns. Despite its ubiquity and global distribution, SAR11 remains incredibly difficult to culture and also creates problems reconstructing MAGs from environmental surveys. As such, this next paper attempts to investigate the genomic diversity of the SAR11 clade using read recruitment of oceanic samples to identify single-amino acid variants [3]. One issue I keep coming across in any investigation into the SAR11 clade is that most studies are examining genetic diversity across an entire order. Is it really surprising that different families, genera, species, populations (if we can get to this level) within the SAR11 order have differential distributions? And why is it applicable to then look at non-synonymous variation? In any case, the authors further extrapolate amino acid variants to physico-chemical properties to conclude that the hydrophobic interactions of amino acids are under purifying selection. Another question I had how many amino acid positions would be affected by the chemistry, is this restricted to the active sites? How did they tease a part neutral divergence in these proteins across divergent taxa? Finally, the paper concludes that proteotypes within a SAR11 subclade exhibit differential biogeographic patterns with temperature being a driving determinant of spatial partitioning. But then they conclude that "lending credence to the idea for marine systems that 'everything is everywhere but the environment selects'"? - shouldn't these distinct biogeographic patterns reflect the opposite? Regional biogeographic patternsRates of homologous recombination between closely-related strains have largely been inferred from genome sequences, but Potnis et al. utilize in vitro recombinant strains along with wild-type isolates to determine the extent of HR [4]. The authors identified large recombining fragments (upwards of 30kbp) in concentrated genomic islands. Functional annotations of these regions revealed genes related to the transition of pathogenic lifestyles of the plant pathogen.
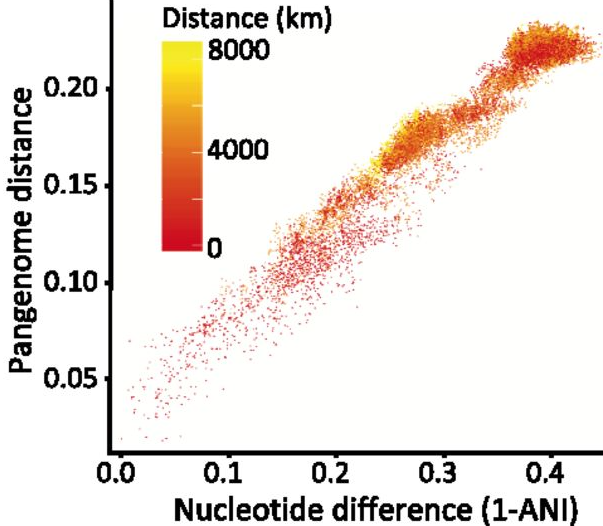
Fine spatial scaleStaying with soil dynamics, next I want to look at this really neat paper that sampled Streptomyces strains at spatial microscales (mm to cm). Similarly to the SAR11 clade, this "genus" encompasses extensive genetic diversity. Based on these ambiguous delineations, I think a lot of studies have overestimated the extent of HGT in structuring Streptomyces diversity; actually a recent study quantified this showing they are indeed rare HGT events across distantly-related Strepto lineages. Back to this paper which isolated 32 strains from soil aggregates that shared >98.6% ANI [6]. Despite this, conspecific strains harbored extensive gene content differences in the flexible genome and proposed that the massive gene fluxes are probably mediated by Actino ICEs (AICEs). Some of these indels included biosynthetic gene clusters (BGCs) - this might only be a cool result for me! In conclusion, the observed gene fluxes between microscale spatially-separated strains was mediated by frequent transfer among closely-related strains via conjugation. This last paper is really interesting as **SPOILER** they show as many as 16?!?!? strains of intracellular symbionts can coexist in a single individual of deep sea mussels [7]. I find this result fascinating since we would expect highly similar strains to outcompete one another via the competitive exclusion principle. In this system, it was thought the mussels hosted two intracellular symbionts, one for sulfur-oxidizing (SOX) and another for methane-oxidizing - both contribute energy sources for carbon fixation. By 16S rRNA standards, each mussel host was thought to harbor a single symbiont species of SOX and methane oxidizers. This paper utilized metagenomes and metatranscriptomes to identify variable strain diversity in a single mussel host. Further, these variations were linked to strain-specific genes related to key functional processes in the SOX symbionts. For instance, some symbiont strains expressed genes related to H2 oxidation; the authors also confirmed with imaging analysis (below). This imaging revealed such fine-scale spatial partitioning that it was restricted to the host bacteriocytes - that is incredible compartmentalization! 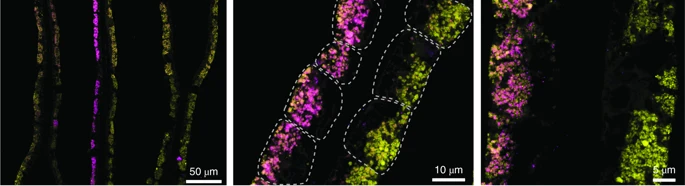 Simultaneous FISH of hydrogenase operon (violet) and 16S rRNA (green) of the SOX symbiont in gill tissue of B. azoricus from site encoding genes related to H2 oxidation. Papers:1. Chase AB, Martiny JBH. (2018). The importance of resolving biogeographic patterns of microbial microdiversity. Microbiology Australia 39(1): 5-8.
2. Greenlon A, Chang PL, Damtew ZM, Muleta A, Carrasquilla-Garcia N, Kim D, Nguyen HP, Suryawanshi V, Krieg CP, Yadav SM, Patel JS, Mukherjee A, Udupa S, Benjelloun, Thami-Alami I, Yasin M, Patil B, Singh S, Sarma BK, von Wettberg EJB, Kahraman A, Bukun B, Assefa F, Tesfaye K, Fikre A, Cook DR. (2019). Global-level population genomics reveals differential effects of geography and phylogeny on horizontal gene transfer in soil bacteria. Proceedings to the National Academy of Sciences 116(30): 15200-15209. 3. Delmont TO, Kiefl E, Kilinc O, Esen OC, Uysal I, Rappe MS, Giovannoni S, Eren AM. (2019). Single-amino acid variants reveal evolutionary processes that shape the biogeography of a global SAR11 subclade. eLife 8:e46497. 4. Potnis N, Kandel PP, Merfa MV, Retchless AC, Parker JK, Stenger DC, Almeida RPP, Bergsma-Vlami M, Westenberg M, Cobine PA, de la Fuente L. (2019). Patterns of inter- and intrasubspecific homologous recombination inform eco-evolutionary dynamics of Xylella fastidosa. The ISME Journal 13: 2319–2333. 5. Chase AB, Arevalo P, Brodie EL, Polz MF, Karaoz U, Martiny JBH. (2019). Maintenance of sympatric and allopatric populations in free-living terrestrial bacteria. mBio 10(5): e02361-19. 6. Tidjani A, Lorenzi J, Toussaint M, van Dijk E, Naquin D, Lespinet O, Bontemps C, Leblond P. (2019). Massive gene flux drives genomic diversity between sympatric Streptomyces conspecifics. mBio 10(5): e01533-19. 7. Ansorge R, Romano S, Sayavedra L, Porras MAG, Kupczok A, Tegetmeyer HE, Dubilier N, Peterson J. (2019). Functional diversity enables multiple symbiont strains to coexist in deep-sea mussels. Nature Microbiology What is the biological question???I wanted to make a blog post about my recent microbiome workshop I led at Scripps Institution of Oceanography this week. The audience was mostly graduate students and faculty in my department whose research focuses on the discovery of natural products from marine organisms (see my previous post here about some cool research). I thought a lot of people might find this useful, especially earlier graduate students interested in microbiome work. So without further ado, here is a rundown of the workshop, including a nice dataset and some workflows I modified from the UCI Microbiome Initiative - special thanks to Julio and Claudia from the Whiteson and Martiny labs. Nowadays everyone is interested in how the microbiome affects [fill in your system here]. This had led to many studies including microbiome community analyses in their experimental design. Sometimes, the analysis does not coincide with the biological question (i.e., species level differentiation using 16S rRNA sampling). My point is to think carefully about what your biological question is and then design the correct sampling method to ensure the data can be interpreted and address your question. Also, there is a lot of limitations with each sequencing method (metagenomics v. amplicon-based) and understanding these limitations is paramount. I wrote another blogpost going over some of these topics.  Briefly, it comes down to the degree of genetic resolution you want in your samples. For instance, the 16S rRNA gene has been instrumental in our understanding of microbial diversity. At the same time, it is a highly conserved marker gene. This means it cannot determine or resolve genetic (and definitely not functional) diversity of closely-related bacteria. “one limitation of the 16S rRNA gene is that it is rather conserved and hence is NOT reliable for taxonomic identifiers at the species level” -J. Cole et al. 2010.
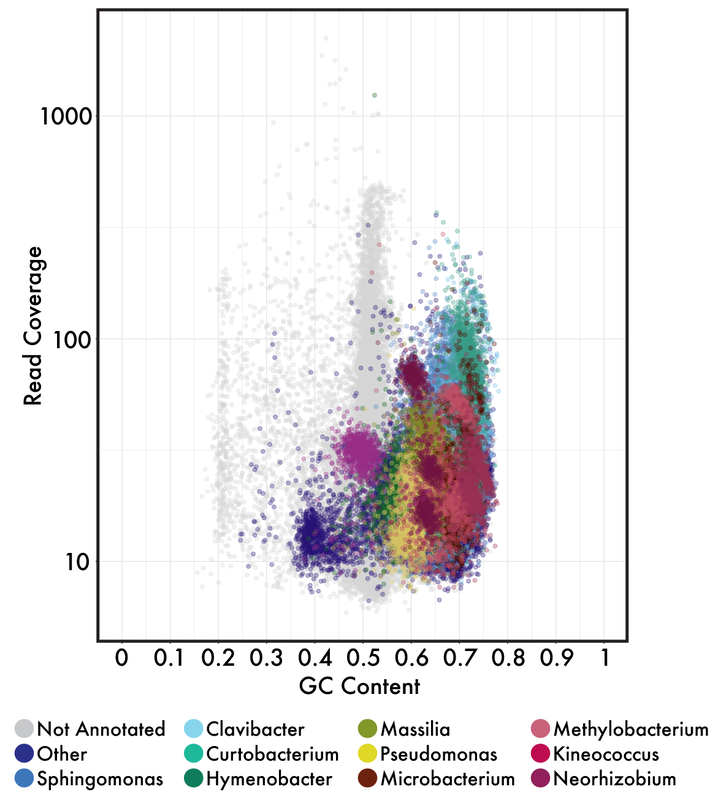
Microbiome DataWhen you sequence a sample, you will get .fastq files or fasta files with quality scores at each base pair position (see powerpoint in github link below). These files will not only give you the DNA sequence information but also information on your sequencing run (e.g., how good is your data). This is where the first couple steps come into play. Whether you have 16S or metagenomic data, you will want to demultiplex your samples (assuming you ran a bunch of samples in a pooled library) and then denoise them. This means you will use those quality scores to trim your reads at a certain threshold. You will also want to trim the adapter sequences and/or primers off the beginning of each read. These steps can easily be done with tons of software - my personal favorite is BBMap but almost all of them will follow similar steps. 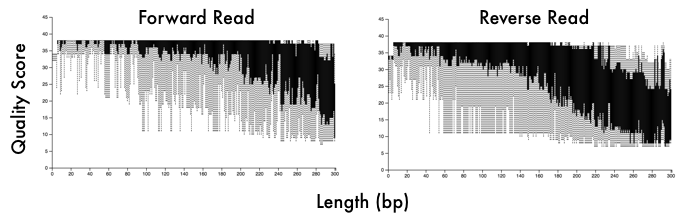 Typical visualization of QC (quality scores) of the fastq files. This data is 16S reads and we can see the adapters in the first 5 bp positions and then see a drastic decrease in quality scores at ~200bp for the forward reads and ~160bp for reverse reads 16S v Metagenomic OptionsMetagenomic Analysis So after you have your "cleaned up" data, what now? Well if you have metagenomic reads, you can perform assemblies and try to bin MAGs (metagenome assembled genomes - see post here). Or you can do a "read-based" approach to characterize the taxonomic diversity (like using MIDAS) or the functional diversity by searching for genes of interest. The latter, read-based approach, is nearly impossible to link function to taxonomy but can give you community-wide aggregated functional diversity. Metagenomic analyses can get really complicated so I will not go further into it here, but if interested here are some really useful links: Anvi'o by the Meren Lab Multi-Metagenome - R-based MAGs pipeline In general, you will need to follow these steps so learning how to navigate in a terminal is pretty essential. Here is some advice from a graduate student, Tyler Barnum at Berkeley. 1. Read assembly (MEGAHIT, metaSPAdes) 2. Read Mapping for coverage profiles (bowtie2, bwa, BBMap) 3. Binning 4. Bin Curation and Quality Check 16S rRNA amplicons This is where I wanted to focus this workshop. For one, I think most people will be analyzing this type of data. Furthermore, it is a lot more "user-friendly" to parse out this data. A ton of computational tools are available to analyze amplicon-based datasets, including QIIME2, DADA2, mothur, usearch, etc. each with tons of community support and tutorials. Post filtering and denoising, you will generally want to 1. Cluster sequences into operational taxonomic units (OTUs) 2. Create an OTU table with abundances of taxa across samples 3. Interpret data Most of the tools available have step-by-step instructions on how to navigate through data processing. These steps are pretty straight-forward and I included an example run in the github link below. One of the major decisions you will need to make during this process is whether to use traditionally-defined OTUs (97% OTUs) or use the growing application of single nucleotide variants (ESVs or ASVs or 100% OTUs). Either way, remember that the 16S has limited resolution and whatever clustering threshold you use cannot give you species level resolution nor intra-species patterns or processes. Don't make this mistake! In any case, your clustering threshold has a lot of debate and back-and-forth in the community so I will refrain from going into this as I am not typically analyzing 16S rRNA data. However, I definitely recommend you reading some information on this and educate yourself on the literature and the reasoning behind each argument. Workshop MaterialsAlright - that is enough background information. Here is the link to the workshop materials. I wanted to use R to analyze data as I think walking through this yourself gives a better understanding of what is happening. R has several advantages including reproducibility and many packages created specifically for microbiome analysis (e.g., phyloseq).
Github link: https://github.com/alex-b-chase/16S-workshop
Introduction to Pangenomes
I wanted to discuss some broad discussion points with bacterial pangenomes and their genetic diversity at large. A lot of these ideas I have been discussing with the amazing microbial ecologist, Linh Anh Cat, Ph.D., and I would definitely recommend following her pieces she writes for Forbes highlighting some really interesting biological findings, including glow in the dark fungi!?
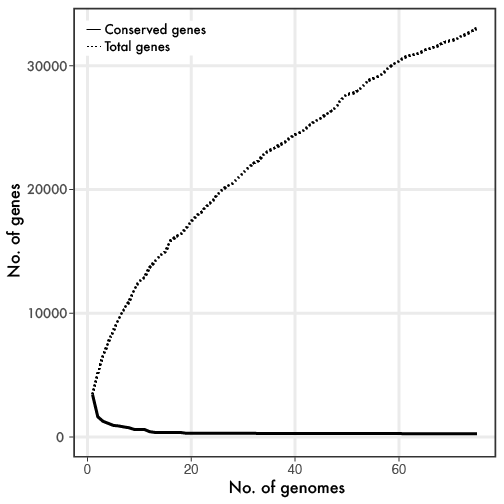
Linh Anh Cat: [Twitter link][Forbes link] With the enormous progress in sequencing microbial genomes, a general phenomenon quickly emerged revealing extensive genetic diversity in genome content, even within closely-related taxonomic groups. The utilization of the core (genes found in all members of a group) and pangenome (all genes found within a group) framework provided a baseline to examine the almost infinite genetic diversity. If you are reading this blog, you have probably seen something similar to the below figure, where the more genomes that are sampled, the smaller the core genome gets while the flexible or pangenome continually increases.
So why do prokaryotes have pangenomes?
A paper [2] a couple of years ago attempted to answer this exact question. By doing so, the authors incorporate a theoretical approach to include effective population sizes, mutation rates, selection coefficients, etc and applied neutral, deleterious, and adaptive models. They concluded that the ability of bacteria to migrate to new micro-niches enable the expansion of the pangenome. Restricted taxa (e.g., obligate intracellular organisms) have largely reduced pangenomes while free-living bacteria can have massive pangenomes. These conclusions sparked a lot of interest especially since a contemporary paper argued the complete opposite, pangenomes are directed by neutral evolution [3].
A response by BJ Shaprio [4] brought up a great point that population-level theory is tough to apply to microbial pangenomes, as there are no clear delineations of where to draw your "cut-off" (see why I thought the original tweet was so great!). Mainly, if pangenomes are driven by HGT, then transfers occur across populations, species, and broader taxonomic boundaries. One can argue for genetic relatedness and/or ecological approaches to accurately delineate where to assess pangenome composition. In the end, the paradox remained opened for debate as large effective population sizes and selection coefficients correlate both with large pangenome sizes. Why is this so interesting? For one, IF flexible genes were advantageous, we would expect selective sweeps across populations within species, thereby reducing pangenome sizes [5]. Disentangling environmental and phylogenetic constraints
So why am I bringing this all up now? Well, two recent preprints were published bringing this debate back into the forefront. To recap, the pangenome can be shaped both by habitat (via selection) and by phylogeny (via vertical inheritance). For instance, more closely-related strains will share more similar genes. However, it is really difficult to separate these two factors, phylogeny and habitat, as more closely-related strains not only share more genes, but should also prefer more similar habitats. The first of the preprint I want to highlight seeks to disentangle these effects and characterize their impact on bacterial pangenomes [6]. Using a large collection of species pangenomes (N=155 species), the authors conclude that the adaptiveness of pangenomes is partially explained by the environmental habitat and their shared core genome.
The second paper uses a more theoretical approach, a model simulation of gene content of pangenomes [7]. What I like most in this paper is that it really tried to resolve the pangenome paradox. Briefly, it comes down to the most biological conclusion, it depends. Large pangenomes of low-frequency genes are neutral. Highly beneficial genes in the pangenome can arise as a consequence of genotype-by-environment when multiple niches are available. And all if this can be influenced by the rate of gene gain and loss. In the end, we need empirical data! 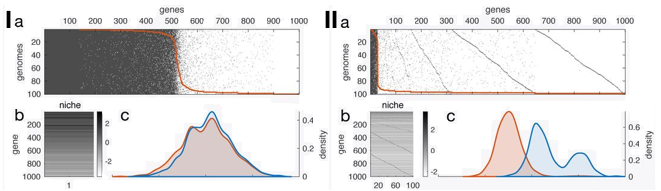
I) Simulated set of 100 genomes from a single niche (1000 genes with varying fitness effects). II) Simulated set of 1 genome from 100 niches with 100 mostly-deleterious genes. A) P/A of genes in simulated pangenome B) Heatmap of sampled fitness effects C) Density plots of gene pool for theoretical (blue) and fitness effect of genes in pangenome (orange)
Papers:
1. Rocha EPC. (2018). Neutral Theory, microbial practice: Challenges in bacterial population genetics. Molecular Biology and Evolution 35(6): 1338-1347.
2. McInerney JO, McNally A, O'Connell MJ. (2017). Why prokaryotes have pangenomes. Nature Microbiology 2: 17040. 3. Andreani NA, Hesse E, Vos M. (2017). Prokaryote genome fluidity is dependent on effective population size. The ISME Journal 11: 1719-1721. 4. Shapiro BJ. (2017). The population genetics of pangenomes. Nature Microbiology 2: 1574. 5. McInerney JO, McNally A, O'Connell MJ. (2017). Reply to 'The population genetics of pangenomes'. Nature Microbiology 2:1575. 6. Maistrenko OM, Mende DR, Luetge M, Hildebrand F, Schmidt TSB, Li SS, Coelho LP, Huerta-Cepas J, Sunagawa S, Bork P. (2019). Disentangling the impact of environmental and phylogenetic constraints on prokaryotic strain diversity. bioRxiv 7. Domingo-Sananes MR, McInerney JO. (2019). Selection-based model of prokaryote pangenomes. bioRxiv Theme of the month: microbes to ecosystem functioningI wanted to get back to some of main goals in microbial ecology: relating microbial composition (or changes in composition) to function. This part of microbial ecology can lean heavily on the work done in community ecology from macroorganisms. In particular, there is a need to move beyond describing patterns of species richness and diversity to get at the functional consequences of this variation across spatiotemporal scales. This allows the use of a trait-based framework to really link traits to ecosystems and to link them inevitably to measures of diversity. As a primer, I would highly recommend reading this book chapter by Enquist et al (2013) which provides a thorough read through of trait based frameworks, including its influences from Grime's Mass Ratio and Metabolic Scaling Theory - both of which can be applied to microbial systems. Without further ado, here are some recent papers tackling this topic: Linking Microbial Diversity to FunctioningTraits underlie an organism's response to both biotic and abiotic factors. These traits will underline a particular organism's geographic distribution. For the most part, studies infer traits from microbial surveys, but linking these remain incredibly difficult. For one, it's impossible to quantify every trait for every microbes in a system. Of course, we can borrow from community ecology and concentrate on the more abundant, dominant members in a system (e.g., some of my work on Curtobacterium [1]). But this requires a lot of work, from isolation, characterization, developing relevant assays, collecting geographic distribution data, etc. Much more feasible is assaying for community-wide "traits" or determining function from aggregated functional responses (e.g., respiration). These approaches should allow for the formulation of hypotheses on microbial trait responses (for more see these two recent reviews here [2,3,4]). Environmental Gradients
Plants to soil to ecosystemsGoing to finish off with 3 papers I liked this past year or so. They are all related to soil microbiomes so sorry for any marine or human microbiome people out there! Do plants, bacteria, and fungi respond similarly by the same environmental variables? A recent study applies trait-based ecology to investigate the effects of temperature [8]. Specifically, should shifts in temperature correspond to shifts in plant traits and microbial function? And are there ecological feedbacks between plants and their microbes? Speaking of the soil microbiome affecting plant health. Wei et al. found that small variation in the initial soil microbiome can affect the health of plants throughout their experiment [9]. These results highlight the plant disease dynamics can be driven by highly deterministic processes based on the microbial composition and functional properties. Lastly, plants can regulate their rhizosphere microbial community by releasing chemical exudates from their roots [10]. The release of these exudates follows a "chemical succession" of microbes driving community assembly. Papers:1. Chase AB, Gomez-Lunar Z, Lopez AE, Li J, Allison SD, Martiny AC, Martiny JBH. (2018). Emergence of soil bacterial ecotypes along a climate gradient. Environmental Microbiology 20: 4112-4126.
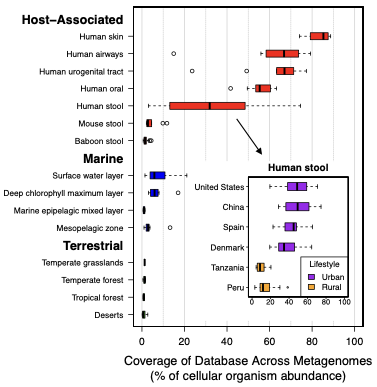
2. Lajoie G, Kembel SW. (2019). Making the most of trait-based approaches for microbial ecology. Trends in Microbiology 27: 814-823. 3. Wang JT, Egidi E, Li J, Singh BK. (2019). Linking microbial diversity with ecosystem functioning through a trait framework. Journal of Biosciences 44: 109. 4. Malik AA, Martiny JBH, Brodie EL, Martiny AC, Treseder KK, Allison SD. (2019). Defining trait-based microbial strategies with consequences for soil carbon cycling under climate change. The ISME Journal 1-9. 5. McGill BJ, Enquist BJ, Weiher E, Westoby M. (2006). Rebuilding community ecology from functional traits. Trends in Ecology and Evolution 4: 178-185. 6. Glassman SI, Weihe C, Li J, Albright MBN, Looby CI, Martiny AC, Treseder KK, Allison SD, Martiny JBH. (2018). Decomposition responses to climate depend on microbial community composition. Proceedings to the National Academy of Sciences 115(47): 11994-11999. 7. Rath KM, Maheshwari A, Rousk J. Linking microbial community structure to trait distributions and functions using salinity as an environmental filter. mBio 10(4): e01607-19. 8. Buzzard V, Michaletz ST, Deng Y, He Z, Ning D, Shen L, Tu Q, Van Nostrand JD, Vooreckers JW, Wang J, Weiser MD, Kaspari M, Waide RB, Zhou J, Enquist BJ. (2019). Continental scale structuring of forest and soil diversity via functional traits. Nature Ecology and Evolution 3: 1298-1308. 9. Wei Z, Gu Y, Friman VP, Kowalchuk GA, Xu Y, Shen Q, Jousset A. (2019). Initial soil microbiome composition and functioning predetermine future plant health. Science Advances 5(9): eaaw0759. 10. Zhalnina K, Louie KB, Hao Z, Mansoori N, da Rocha UN, Shi S, Cho H, Karaoz U, Loque D, Bowen BP, Firestone MK, Northen TR, Brodie EL. (2018). Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nature Microbiology 3: 470-480. Recently, a lot of conversation has been circulating regarding what constitutes a microbe. How do we know we captured the diversity in a system and have a representative sample or sequence? Are abundant, relevant members of a community culturable? If not, how can we be sure that the computational output is representative? Especially with the increased use of culture-independent methods in microbiome research, we are relying heavily on the molecular and computational methods to assess this vast diversity of microorganisms. For this post, I will focus less on ideas of microdiversity (although this will come briefly later) within 16S defined OTU/ESVs. Instead, I want to highlight some of the more interesting conversations happening in microbial ecology regarding 1) culturability and 2) metagenome-assembled genomes or MAGs. This post largely spawned from the amazing dialogue being generated over Twitter over these two issues, and I just wanted to give my opinion on the topics. I will refrain from "taking sides" but focus on the arguments and the bigger picture implications for microbiome research. High proportions of bacteria are culturable across major biomesLet's start here because I think this is a SUPER important topic and obviously will relate to the second conversation below. For decades, microbiologists have leaned heavily on a major crutch: paradigm that only 1% of microorganisms are culturable A recent communication by Adam Martiny in ISME [1] challenged this paradigm in microbiology by arguing that, based on 16S rRNA similarity of bacteria, abundant members (35-52% of sequences and taxa, respectively) across major biomes have a representative member in a cultured isolate. At first, I was delighted to see this communication published. Just weeks before this, I saw multiple seminars and job talks starting the Introduction with highlighting this paradigm. Inevitably, it came across, that "well we can't culture bacteria, so why even try?". Subsequently, the talks would go over culture-independent approaches to navigate this "massive hurdle" in microbiology. But this is a major disservice to all the hardwork done to culture bacteria. Everything, in theory, is culturable - it's a matter of figuring out how to do it. The question shouldn't be is X bacterium culturable? it should be how do we culture it? This ideology of yet-to-be cultured microbes is definitely painstaking work, but it is possible (everyone please check out the incredible work of Yoichi Kamagata to characterize anaerobic bacteria and their syntrophic relationships - some culturing issues were resolved just by switching the preparation of the media! [2]).
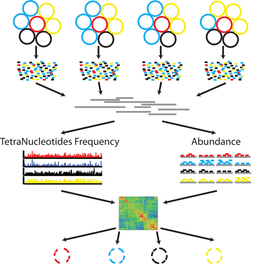
Both of these papers make great points and address topics that every microbiologists should be thinking about. As I have said before, many studies infer far too much from descriptive datasets of 16S rRNA community analyses. It is almost impossible to relate this data or the observed taxa to ecological function. I have spent a lot of time laying out some of these arguments, but basically comes down to the 16S rRNA gene provides a coarse depiction of the microbial community. There are millions of years of divergence and adaptation masked by the use of conserved marker genes. These are some of the similar points the Steen et al. paper lay out to argue against the idea that most organisms have been cultured. However, I do feel like this resolution, specifically with 16S rRNA studies, it is relevant to compare against databases. This is exactly what Martiny's response [4] details that the most common interpretation using a defined taxonomic unit (OTU) implies a lot of abundant members have cultured representatives in databases, certainly superseding the 1% "rule". In any case, how you define whether a taxa is cultured or what is the real number so far really depends on interpretation. And based on this interpretation, researchers need to apply appropriate methods. If you are concerned with fine-scale diversity, do not use 16S rRNA surveys. If you want to target a specific group, design taxa-specific primers. If you want to infer function and avoid PCR and molecular biases, probably switch to metagenomes. And finally, if you wish to link genotype to phenotype, figure out a way to culture and target the abundant members of your system. Physiological data can provide a lot and was the basis for most of my work to target isolates. Artificial construction of metagenome assembled genomes (MAGs)???I think this is a nice segue into the next topic. Based on the vastly different ways to interpret culturability of a microbe, I think it is super interesting that MAGs aren't more of a topic of discussion. For those who are unfamiliar, MAGs are extracted from metagenomic data using computational approaches to assemble "community" data, bin assembled contigs into similar sequences based on genomic signatures (e.g., %GC, tetranucleotide frequencies), and assess contamination to create MAGs of otherwise yet-to-be-cultured ;) bacteria or archaea (schematic below).
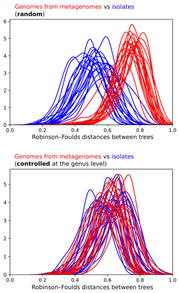
As many members were quick to point out, cultured isolates exist for the novel lineages where high quality MAGs (i.e., complete genomes) match pretty nicely. As an ultra bonus, one of the "artificial" lineages detailed in the preprint recently was isolated and described in another preprint documenting a 12 year(!) isolation effort for Asgard Archaea - this is why science is so awesome! Further, a really talented grad student in the Banfield Lab, Alex Crits-Christoph, re-analyzed this data to address the main support against MAGs in the preprint: The effect observed is due to differences in the phylogenetic distribution of genomes from metagenomes vs isolate genomes.
I personally think the authors vastly overstate their conclusions but I do think they bring up a few good points, especially if we start to bring in the ideas from the previous section. MAGs are useful because we can essentially sequence the entire microbial community and "pull out" genomes without isolating strains. This was incredibly useful in simple microbial communities where low genetic diversity allowed for the relatively easy binning of MAGs. However, I do start to question how much of MAGs, especially in complex, genetically-diverse systems, are a representation of a mosaic of closely-related strains and/or lineages.
Papers:1. Martiny AC. (2019). High proportions of bacteria are culturable across major biomes. ISME 13: 2125-2128.
2. Kato S, Yamagishi A, Daimon S, Kawasaki K, Tamaki H, Kitagawa W, Abe A, Tanaka M, Sone T, Asano K, Kamagata Y. (2018). Isolation of Previously Uncultured Slow-Growing Bacteria by Using a Simple Modification in the Preparation of Agar Media. Applied Environmental Microbiology 84: e00807–18. 3. Steen AD, Crits-Christoph A, Carini P, DeAngelis KM, Fierer N, Lloyd KG, Thrash JC. (2019). High proportions of bacteria and archaea across most biomes remain uncultured. ISME. 4. Martiny AC. (2019). The '1% culturability paradigm' needs to be carefully defined. ISME. 5. Garg SG, Kapust N, Lin W, Tria FDK, Nelson-Sathi S, Gould SB, Fan L, Zhu R, Zhang C, Martin WF. (2019). Anomalous phylogenetic behavior of ribosomal proteins in metagenome assembled genomes. bioRxiv. 6. Chase AB, Karaoz U, Brodie EL, Gomez-Lunar Z, Martiny AC, Martiny JBHM. (2017). Microdiversity of an abundant terrestrial bacterium encompasses extensive variation in ecologically relevant traits. mBio 8: e01809-17. Natural ProductsAbout 9 months ago, I started my postdoc in a brand new field, natural products. Instead of focusing on the processes contributing to microbial diversity and diversification, the objective is to utilize microbes for the discovery of new natural products. As a total novice, I was blown away by the amazing work being done to quantify single microbial compounds. So, as I look back on what I learned over the past few months, I just wanted to highlight some of the amazing and novel techniques being used to give us insights into the structural diversity and complexity of natural products. This is my (very) humble understanding of natural products and definitely going to be a bit biased towards Scripps and marine sediment bacterium!
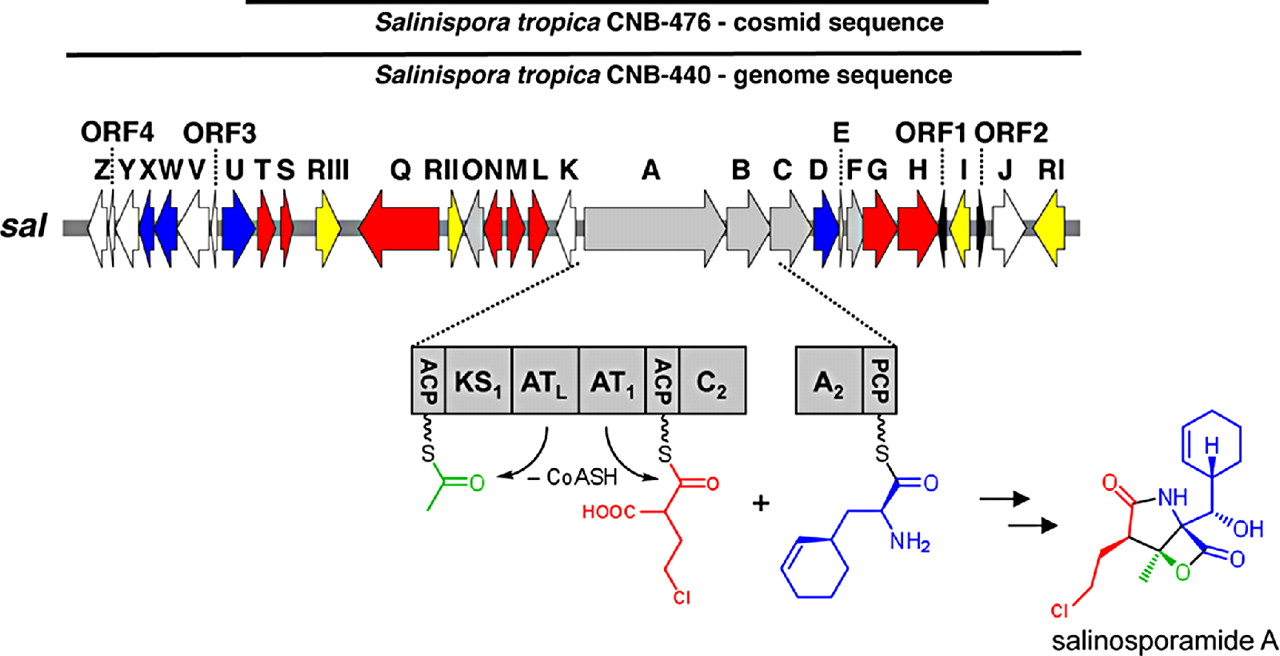
Genome Mining for NPsOf course I would start here! Obviously, the major advances in sequencing technology and costs have allowed for extensive mining of genomes for genetic signatures related to the production of natural products. In bacteria, these genes are typically clustered in the genome to build proteins in a modular fashion (akin to a car assembly line). Modules within these biosynthetic gene clusters (BGCs) enable the loading, attachment, and extension of building blocks to produce NPs (below Figure).
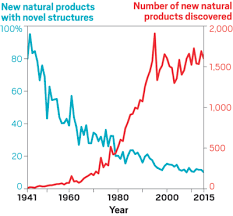
Decades of work identifying novel genomic signatures for BGCs has allowed for extensive surveys across bacterial taxa. Most notably are the Actinobacteria, which include the well-studied NP producing genera such as Streptomyces and Salinispora. Genome mining has revealed large portions of the genome can be dedicated to the production of NPs. This computational approach assesses the entire biosynthetic potential of an organism rather than examining individual metabolites (of which may or may not be expressed in culture conditions due to regulation or environmental signaling). Mass SpectrometryTraditionally, strains are grown in culture and crude extracts are examined to determine which products a bacteria might be producing. This led to the one strain many compounds (OSMAC) approach to characterize diverse compounds in different culture conditions (see below figure). However, analyzing these crude extracts are difficult as secondary metabolites are highly diverse in their size, structure, and physicochemical properties. Instruments such as the LC-MS (liquid chromatography - mass spectrometry) can separate and identify masses of compounds, but still an organisms can produce hundreds of compounds in any given sample. Much work is still to be done, but analytical tools, such as GNPS and MZMine, can aid with the data processing and identification (and dereplication) to characterize compounds. 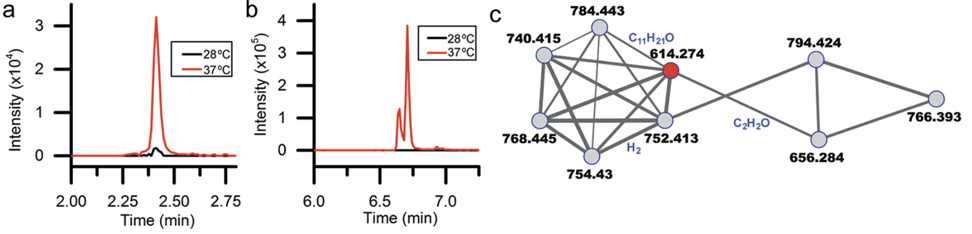 Identification of molecules A) 614.27 m/z and B) 754.44 m/z in high temperature culturing conditions. C) Molecular networking based on MS2 spectra (via GNPS) clustered both masses with a known natural product (red node) [3]. Heterologous Expression of BGCsLinking the identification of BGCs to their products using mass spec is really difficult. As such, most of the "low-hanging fruit" have been characterized in most model organisms for natural products research. This has led to some creative approaches to identify novel secondary metabolites. With the advances in computational tools, exploring biosynthetic potential in the genome has revealed a number of "orphan" BGCs in genomes, or identified BGCs that have yet to be linked to its corresponding molecule.
Papers:1. Pye CR, Bertin MJ, Lokey RS, Gerwick WH, Linington RG. (2017). Retrospective analysis of natural products provides insights for future discovery trends. Proceedings to the National Academy of Sciences 114(22): 5601-5606.
2. Eustáquio AS, McGlinchey RP, Liu Y, Hazzard C, Beer LL, Florova G, Alhamadsheh MM, Lechner A, Kale AJ, Kobayashi Y, Reynolds KA, Moore BS. (2009). Biosynthesis of the salinosporamide A polyketide synthase substrate chloroethylmalonyl-coenzyme A from S-adenosyl-l-methionine. Proceedings to the National Academy of Sciences 106(30): 12295-12300. 3. Sidebottom AM, Johnson AR, Karty JA, Trader DJ, Carlson EE. (2013). Integrated metabolomics approach facilitates discovery of an unpredicted natural product suite from Streptomyces coelicolor M145. ACS Chemical Biology 8: 2009-2016. 4. Zhang JJ, Moore BS, Tang X. (2018). Engineering Salinispora tropica for heterologous expression of natural product biosynthetic gene clusters. Applied Microbiology and Biotechnology 102(19): 8437-8446.
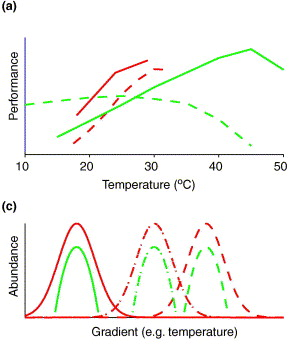
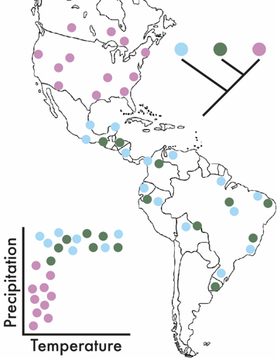
This is where adapting a biogeographic framework can be powerful. It allows us to assess the processes controlling the geographic distribution of species over space and time. It can incorporate ecological and environmental factors (e.g., temperature and precipitation) while also allowing for the integration of phylogeography to assess evolutionary processes (Figure). These ideas have a rich history in plant and animal communities and have lead to the develop of ground-breaking ecological theories attempting to explain species diversity and distribution (for some ecological background, see MacArthur-Wilson, Hubbell's Neutral Theory, and/or a nice synthesis by M Vellend). In the past two decades, these patterns have largely been explored in microbial communities, reflecting the importance of selection of environmental conditions based on correlations between microbial composition and the environment. For instance, one of the first papers to really explore these patterns showed soil bacterial communities were highly influenced by pH [1]. However, there is a large disconnect between theoretical and empirical work conducted in plants and animals to microbes, as the former are studied at the species level and describe large-scale patterns of species’ distributions. By concentrating on finer-genetic resolutions (i.e., species and population levels) we can better detect the eco/evo processes contributing to the maintenance of microbial diversity [2]. Deterministic or Stochastic?!?!For this section, I just want to highlight some processes that have been shown (in either bacteria or macroorganisms) to maintain species diversity. For the ease of this blog post, I will broadly separate these into two major categories, deterministic and stochastic processes. This, in no way comprehensive list is mainly derived from the amazing ecology grad course taught by my PhD advisor, Jennifer Martiny at UC Irvine.
This is where I think microbial ecologists can vastly expand our understanding of the tremendous microbial diversity. For instance, niche partitioning requires relating traits to phylogeny which should reflect differential environmental distributions. Often, this requires quantifying traits of closely-related bacteria as the competitive exclusion principle predicts a limitation to niche differences. Furthermore, huge efforts are being made to quantify the relative impacts of deterministic v. stochastic processes in microbial studies, both from theoretical frameworks [3] to experimental [4]. Papers:1. Fierer N & Jackson RB. (2006). The diversity and biogeography of soil bacterial communities. Proceedings to the National Academy of Sciences 103(3): 626-631.
2. Chase AB & Martiny JBH. (2018). The importance of resolving biogeographic patterns of microbial microdiversity. Microbiology Australia 39(1): 5-8. 3. Ning D, Deng Y, Tiedje JM, Zhou J. (2019). A general framework for quantitatively assessing ecological stochasticity. Proceedings to the National Academy of Sciences 116(34): 16892-16898. 4. Albright MBN, Chase AB, Martiny JBH. (2019). Experimental evidence that stochasticity contributes to bacterial composition and functioning in a decomposer community. mBio 10:e00568-19. Theme of the month: horizontal gene transferHGT has the power to accelerate evolution by introducing novel alleles across phylogenetically distant taxa. At the same time, rampant HGT can blur species boundaries and is expected to result in a mosaic of genes. I have always been fascinated by the idea that bacteria undergo rampant HGT insomuch that there have been previous proposals for a "web of life". As much as HGT can contribute to rapid diversification, there is also strong evidence for cohesive bacterial lineages, which is fundamental to our understanding of biodiversity in the microbial kingdom. These ideas seem to create an evolutionary "tug-of-war" between local adaptation mediated via acquisition of beneficial alleles and shared phylogenetic history. Here, I just wanted to highlight some new(ish), excellent papers addressing the idea of HGT across multiple scales, from theoretical to empirical, and HGT across kingdoms. Brief Introduction: HGT vs. recombination
At the same time, there are interesting patterns when looking at closely-related bacteria. For one, when comparing genomes of closely-related bacteria, many of the genes in the genomes are unique, or not found in all strains. These genes are collectively referred to the pan-genome. Repeatedly, studies find almost infinite pan-genomes when surveying bacterial groups. Whether these genes are neutral or are a result of frequent adaptive HGT in local environments remains up for debate. Some have proposed that local populations can tap into a shared gene pool as many of these accessory or flexible genes are under frequency-dependent selection [2]. This would suggest that flexible genes are under strong environmental selective pressures. While HGT can possibly provide beneficial fitness effects, there must be a cost to acquiring foreign DNA. Simply put, for adaptive genes to be maintained in a population, the beneficial effects must far out-weigh detrimental effects or genetic drift [3]. This is why HGT fascinates me. There is an intrinsic cost to accepting foreign DNA, but we know HGT can drastically shape microbial evolution. Personally, I believe many studies have overestimated HGT events or are documenting "evolutionary relic" events, such as using gene homology to overestimate the extent of HGT in bacterial genomes. This is why, I suggest when reading the following list to keep in mind some questions: 1. How prevalent is HGT in natural systems? Obviously we can induce genetic exchange in the laboratory or under high selective pressures, but how common are these events in nature? 2. Is HGT a hand-wavy explanation for unexplained genomic patterns? By comparing disparate genomes from across different environments we may be overestimating HGT events and masking the local adaptation of microbial populations. That's enough of my rambling, let's get to the good stuff: Some interesting modes of HGT transferLet's start with phages.
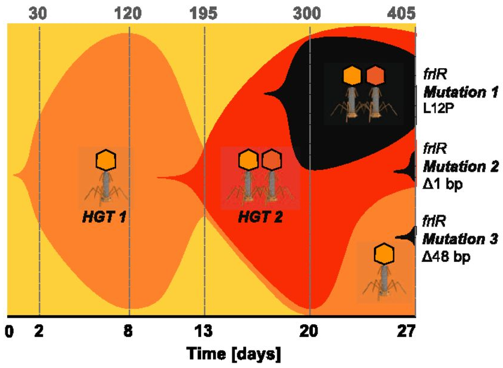
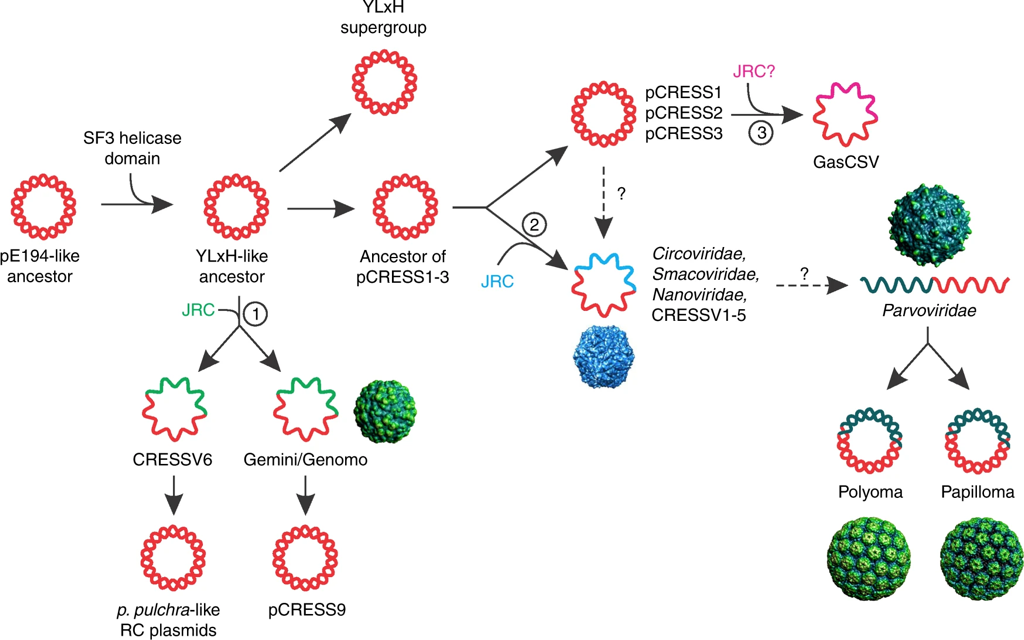
Probably the most studied form of HGT is antibiotic resistance. A couple years ago, a paper from a French research group addressed the acquisition of antibiotic resistance genes mediated through phages. Specifically, this paper addressed the issue in antibiotic resistance gene detection. Inevitably, they find most detected resistance genes were overestimated ("inflated false positives") and that phages rarely encode genes related to antibiotic resistance [5]. The contrast of these first two papers is great! Phages can obviously mediate HGT but the genes being distributed are highly variable. A small shift in gears now. I want to keep discussing phages but move on to the origin of phages. So, for the next paper, we will look at the origin of single-stranded DNA viruses from traditionally exchanged plasmids in bacteria and archaea. ssDNA viruses replicate via the Rep protein of the HUH endonuclease family, a mechanism also found in plasmids. By exploring the relationships among Rep-encoding DNA viruses and transposons from plasmids, the researchers conclude that the origins of ssDNA viruses can be traced to prokaryotic plasmids [6].
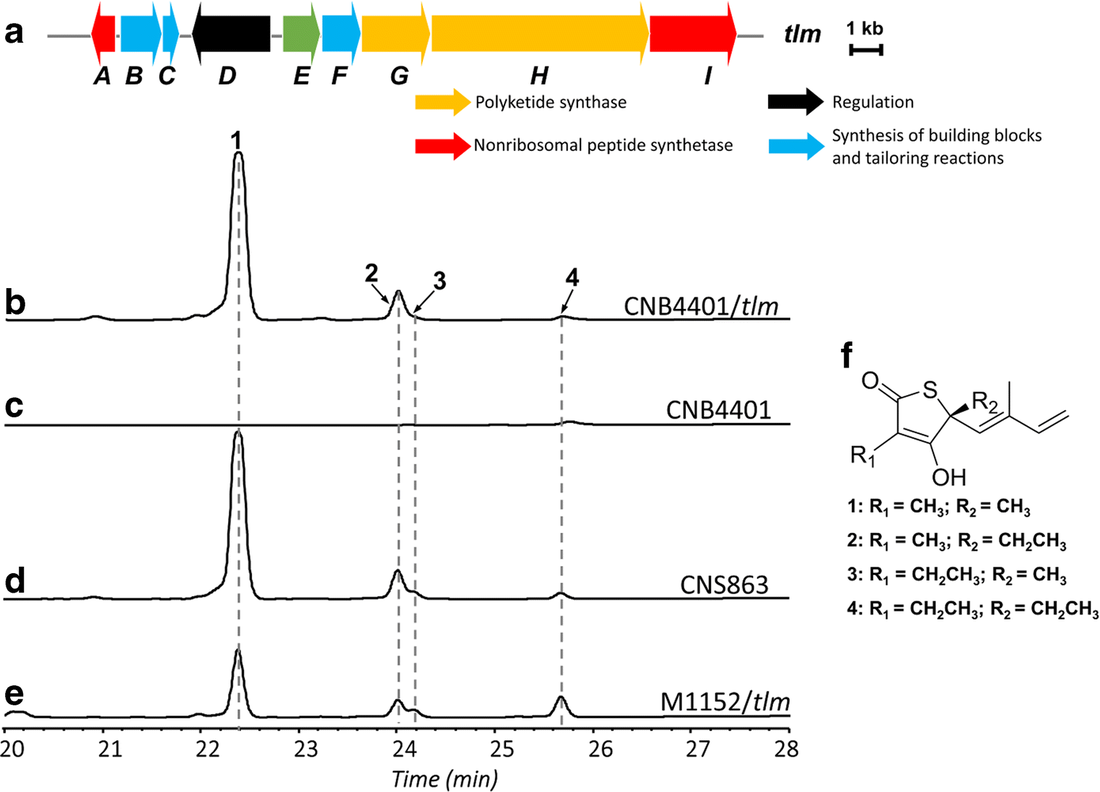
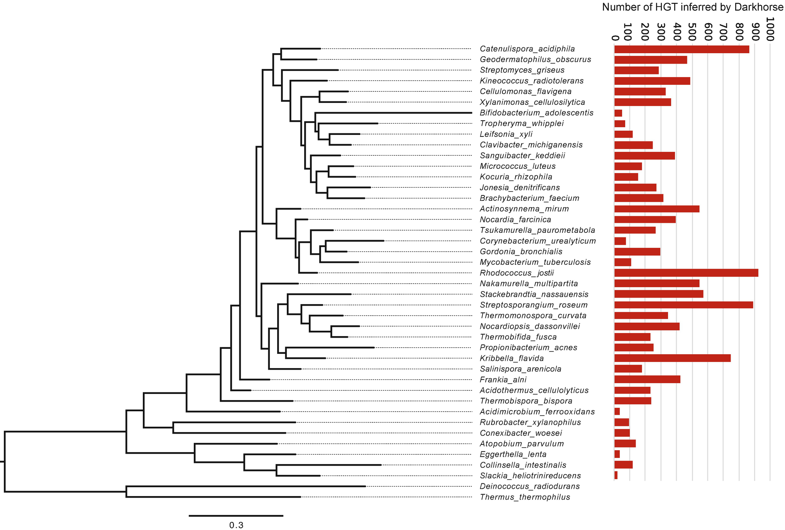
Next up, plamids! Speaking of plasmids, a recent paper documents the presence of a large megaplasmid, with the shared genetic potential to replicate, transcribe, and repair DNA as another closely-related megaplasmid [7]. Megaplasmids, themselves, are evolutionary interesting as the maintenance of these large extra-chromosomal regions possess greater evolutionary costs. The two analyzed in this study suggest strong selective pressures to maintain genetic synteny; while high divergence between orthologous groups suggest independent evolution from a common ancestral plasmid. HGT vectors such as plasmids are undergoing evolutionary processes themselves - they're not just vectors! Cross-kingdom HGTBacterial operons are unique and ubiquitous. They encode the transcription, translation, and production of proteins all in tandem. Bacteria couple these processes by clustering genes into operons, while eukaryotes spatially and temporally separate these processes. These are fundamental differences in metabolism between kingdoms. The next paper, however, found a bacterial operon being transferred, acquired, and maintained in a fungal lineage [8]. After acquisition, the operon underwent structural changes to integrate into eukaryotic synthesis - crazy! The encoded siderophore cluster maintained its high gene clustering in the fungal genome while being modified through transcription including polyadenylation. This paper highlights the boundaries of cross-domain gene transfer for the integration of a complex metabolic pathway. Detection of HGT eventsLastly, I just want to finish with some interesting reports into the frequency of HGT in bacterial systems, specifically in the Actinobacteria phyla. A recent edition on HGT included a detailed account of how HGT can shape evolution in Actinobacteria [9]. These examples include overviews of Streptomyces and Salinispora (very new and dear to me now). In both cases, the authors detail the exchange of large contiguous biosynthetic gene clusters (BGCs) and their relation to HGT, specifically a "plug-and-play" model of evolution where BGCs can be swapped in and out in concentrated genomic islands. To me, this seems extraordinary as HGT should come with (mostly) deleterious fitness costs and the integration of large genomic segments (>30kbp!!!) is hard to wrap my head around. Further, an analysis from the authors inferring HGT events in the Actinobacteria phylum is astounding (below figure), but part of me questions whether this is far overestimating HGT events. Or are we far too liberal with what we classify HGT events? For instance, a paper a couple years ago found that HGT events in Streptomyces were actually quite rare, on the order of 10 genes per million years were acquired and maintained [10]. This would make the transfer of entire BGCs almost unheard of! I would love to hear everyone else's perspective on this front. Again, I am blown away with HGT and really interested in the topic. There are tons and tons of papers I cannot even begin to break down, so let me know your thoughts!
Papers:1. Rocha EPC. (2018). Neutral Theory, microbial practice: challenges in bacterial population genetics. Molecular Biology and Evolution 35: 1338-1347.
2. Polz MF, Alm EJ, Hanage WP. (2013). Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends in Genetics 29: 170-175. 3. Baltrus DA. (2013). Exploring the costs of horizontal gene transfer. Trends in Ecology and Evolution. 28: 489-495. 4. Frazão N, Sousa A, Lässig M, Gordo I. (2019). Horizontal gene transfer overrides mutation in Escherichia coli colonizing the mammalian gut. Proceedings to the National Academy of Sciences 116(36): 17906-17915. 5. Enault F, Briet A, Bouteille L, Roux S, Sullivan MB, Petit MA. (2017). Phages rarely encode antibiotic resistance genes: a cautionary tale for virome analyses. The ISME Journal 11: 237-247. 6. Kazlauskas D, Varsani A, Koonin EV, Krupovic M. (2019). Multiple origins of prokaryotic and eukaryotic single-stranded DNA viruses from bacterial and archaeal plasmids. Nature Communications 10:3425. 7. Smith BA, Leligdon C, Baltrus DA. (2019). Just the two of us? A family of Pseudomonas megaplasmids offers a rare glimpse into the evolution of large mobile elements. Genome Biology Evolution 11(4): 1223-1234. 8. Kominek J, Doering DT, Opulente DA, Shen XX, Zhou X, DeVirgilio J, Hulfachor AB, Groenewald M, Mcgee MA, Karlen SD, Kurtzman CP, Rokas A, Hittinger CT. (2019). Eukaryotic acquisition of a bacterial operon. Cell 176: 1356-1366. 9. Park CJ, Smith JT, Andam CP. (2019). Horizontal gene transfer and genome evolution in the phylum Actinobacteria. In: Villa T., Viñas M. (eds) Horizontal Gene Transfer. Springer, Cham 10. McDonald BR, Currie CR. (2017). Lateral gene transfer dynamics in the ancient bacterial genus Streptomyces. mBio 8: e00644-17. |
AuthorSome thoughts on some (small) things Archives
May 2023
Categories |

 RSS Feed
RSS Feed