What is the biological question???I wanted to make a blog post about my recent microbiome workshop I led at Scripps Institution of Oceanography this week. The audience was mostly graduate students and faculty in my department whose research focuses on the discovery of natural products from marine organisms (see my previous post here about some cool research). I thought a lot of people might find this useful, especially earlier graduate students interested in microbiome work. So without further ado, here is a rundown of the workshop, including a nice dataset and some workflows I modified from the UCI Microbiome Initiative - special thanks to Julio and Claudia from the Whiteson and Martiny labs. Nowadays everyone is interested in how the microbiome affects [fill in your system here]. This had led to many studies including microbiome community analyses in their experimental design. Sometimes, the analysis does not coincide with the biological question (i.e., species level differentiation using 16S rRNA sampling). My point is to think carefully about what your biological question is and then design the correct sampling method to ensure the data can be interpreted and address your question. Also, there is a lot of limitations with each sequencing method (metagenomics v. amplicon-based) and understanding these limitations is paramount. I wrote another blogpost going over some of these topics. 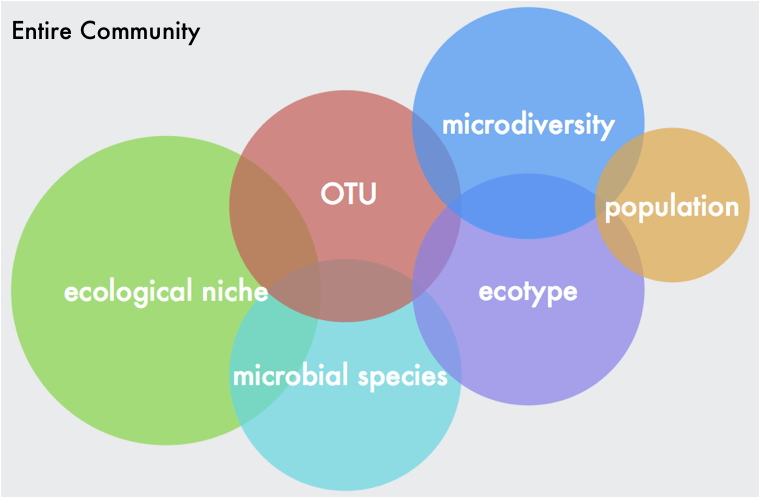 Briefly, it comes down to the degree of genetic resolution you want in your samples. For instance, the 16S rRNA gene has been instrumental in our understanding of microbial diversity. At the same time, it is a highly conserved marker gene. This means it cannot determine or resolve genetic (and definitely not functional) diversity of closely-related bacteria. “one limitation of the 16S rRNA gene is that it is rather conserved and hence is NOT reliable for taxonomic identifiers at the species level” -J. Cole et al. 2010.
Microbiome DataWhen you sequence a sample, you will get .fastq files or fasta files with quality scores at each base pair position (see powerpoint in github link below). These files will not only give you the DNA sequence information but also information on your sequencing run (e.g., how good is your data). This is where the first couple steps come into play. Whether you have 16S or metagenomic data, you will want to demultiplex your samples (assuming you ran a bunch of samples in a pooled library) and then denoise them. This means you will use those quality scores to trim your reads at a certain threshold. You will also want to trim the adapter sequences and/or primers off the beginning of each read. These steps can easily be done with tons of software - my personal favorite is BBMap but almost all of them will follow similar steps. 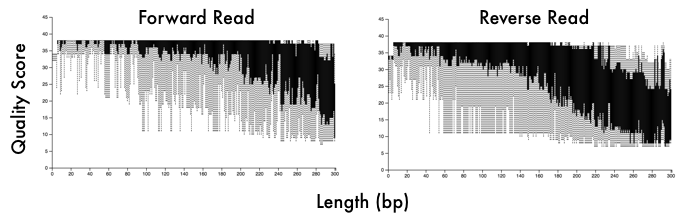 Typical visualization of QC (quality scores) of the fastq files. This data is 16S reads and we can see the adapters in the first 5 bp positions and then see a drastic decrease in quality scores at ~200bp for the forward reads and ~160bp for reverse reads 16S v Metagenomic OptionsMetagenomic Analysis So after you have your "cleaned up" data, what now? Well if you have metagenomic reads, you can perform assemblies and try to bin MAGs (metagenome assembled genomes - see post here). Or you can do a "read-based" approach to characterize the taxonomic diversity (like using MIDAS) or the functional diversity by searching for genes of interest. The latter, read-based approach, is nearly impossible to link function to taxonomy but can give you community-wide aggregated functional diversity. Metagenomic analyses can get really complicated so I will not go further into it here, but if interested here are some really useful links: Anvi'o by the Meren Lab Multi-Metagenome - R-based MAGs pipeline In general, you will need to follow these steps so learning how to navigate in a terminal is pretty essential. Here is some advice from a graduate student, Tyler Barnum at Berkeley. 1. Read assembly (MEGAHIT, metaSPAdes) 2. Read Mapping for coverage profiles (bowtie2, bwa, BBMap) 3. Binning 4. Bin Curation and Quality Check 16S rRNA amplicons This is where I wanted to focus this workshop. For one, I think most people will be analyzing this type of data. Furthermore, it is a lot more "user-friendly" to parse out this data. A ton of computational tools are available to analyze amplicon-based datasets, including QIIME2, DADA2, mothur, usearch, etc. each with tons of community support and tutorials. Post filtering and denoising, you will generally want to 1. Cluster sequences into operational taxonomic units (OTUs) 2. Create an OTU table with abundances of taxa across samples 3. Interpret data Most of the tools available have step-by-step instructions on how to navigate through data processing. These steps are pretty straight-forward and I included an example run in the github link below. One of the major decisions you will need to make during this process is whether to use traditionally-defined OTUs (97% OTUs) or use the growing application of single nucleotide variants (ESVs or ASVs or 100% OTUs). Either way, remember that the 16S has limited resolution and whatever clustering threshold you use cannot give you species level resolution nor intra-species patterns or processes. Don't make this mistake! In any case, your clustering threshold has a lot of debate and back-and-forth in the community so I will refrain from going into this as I am not typically analyzing 16S rRNA data. However, I definitely recommend you reading some information on this and educate yourself on the literature and the reasoning behind each argument. Workshop MaterialsAlright - that is enough background information. Here is the link to the workshop materials. I wanted to use R to analyze data as I think walking through this yourself gives a better understanding of what is happening. R has several advantages including reproducibility and many packages created specifically for microbiome analysis (e.g., phyloseq).
Github link: https://github.com/alex-b-chase/16S-workshop
0 Comments
Introduction to Pangenomes
I wanted to discuss some broad discussion points with bacterial pangenomes and their genetic diversity at large. A lot of these ideas I have been discussing with the amazing microbial ecologist, Linh Anh Cat, Ph.D., and I would definitely recommend following her pieces she writes for Forbes highlighting some really interesting biological findings, including glow in the dark fungi!?
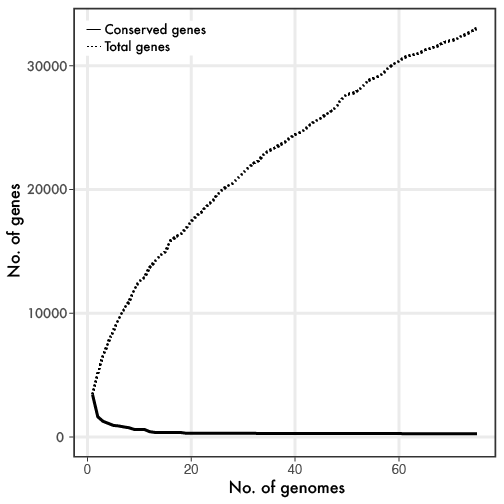
Linh Anh Cat: [Twitter link][Forbes link] With the enormous progress in sequencing microbial genomes, a general phenomenon quickly emerged revealing extensive genetic diversity in genome content, even within closely-related taxonomic groups. The utilization of the core (genes found in all members of a group) and pangenome (all genes found within a group) framework provided a baseline to examine the almost infinite genetic diversity. If you are reading this blog, you have probably seen something similar to the below figure, where the more genomes that are sampled, the smaller the core genome gets while the flexible or pangenome continually increases.
So why do prokaryotes have pangenomes?
A paper [2] a couple of years ago attempted to answer this exact question. By doing so, the authors incorporate a theoretical approach to include effective population sizes, mutation rates, selection coefficients, etc and applied neutral, deleterious, and adaptive models. They concluded that the ability of bacteria to migrate to new micro-niches enable the expansion of the pangenome. Restricted taxa (e.g., obligate intracellular organisms) have largely reduced pangenomes while free-living bacteria can have massive pangenomes. These conclusions sparked a lot of interest especially since a contemporary paper argued the complete opposite, pangenomes are directed by neutral evolution [3].
A response by BJ Shaprio [4] brought up a great point that population-level theory is tough to apply to microbial pangenomes, as there are no clear delineations of where to draw your "cut-off" (see why I thought the original tweet was so great!). Mainly, if pangenomes are driven by HGT, then transfers occur across populations, species, and broader taxonomic boundaries. One can argue for genetic relatedness and/or ecological approaches to accurately delineate where to assess pangenome composition. In the end, the paradox remained opened for debate as large effective population sizes and selection coefficients correlate both with large pangenome sizes. Why is this so interesting? For one, IF flexible genes were advantageous, we would expect selective sweeps across populations within species, thereby reducing pangenome sizes [5]. Disentangling environmental and phylogenetic constraints
So why am I bringing this all up now? Well, two recent preprints were published bringing this debate back into the forefront. To recap, the pangenome can be shaped both by habitat (via selection) and by phylogeny (via vertical inheritance). For instance, more closely-related strains will share more similar genes. However, it is really difficult to separate these two factors, phylogeny and habitat, as more closely-related strains not only share more genes, but should also prefer more similar habitats. The first of the preprint I want to highlight seeks to disentangle these effects and characterize their impact on bacterial pangenomes [6]. Using a large collection of species pangenomes (N=155 species), the authors conclude that the adaptiveness of pangenomes is partially explained by the environmental habitat and their shared core genome.
The second paper uses a more theoretical approach, a model simulation of gene content of pangenomes [7]. What I like most in this paper is that it really tried to resolve the pangenome paradox. Briefly, it comes down to the most biological conclusion, it depends. Large pangenomes of low-frequency genes are neutral. Highly beneficial genes in the pangenome can arise as a consequence of genotype-by-environment when multiple niches are available. And all if this can be influenced by the rate of gene gain and loss. In the end, we need empirical data! 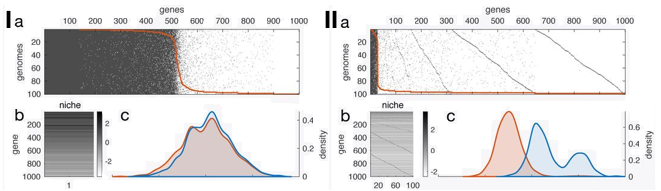
I) Simulated set of 100 genomes from a single niche (1000 genes with varying fitness effects). II) Simulated set of 1 genome from 100 niches with 100 mostly-deleterious genes. A) P/A of genes in simulated pangenome B) Heatmap of sampled fitness effects C) Density plots of gene pool for theoretical (blue) and fitness effect of genes in pangenome (orange)
Papers:
1. Rocha EPC. (2018). Neutral Theory, microbial practice: Challenges in bacterial population genetics. Molecular Biology and Evolution 35(6): 1338-1347.
2. McInerney JO, McNally A, O'Connell MJ. (2017). Why prokaryotes have pangenomes. Nature Microbiology 2: 17040. 3. Andreani NA, Hesse E, Vos M. (2017). Prokaryote genome fluidity is dependent on effective population size. The ISME Journal 11: 1719-1721. 4. Shapiro BJ. (2017). The population genetics of pangenomes. Nature Microbiology 2: 1574. 5. McInerney JO, McNally A, O'Connell MJ. (2017). Reply to 'The population genetics of pangenomes'. Nature Microbiology 2:1575. 6. Maistrenko OM, Mende DR, Luetge M, Hildebrand F, Schmidt TSB, Li SS, Coelho LP, Huerta-Cepas J, Sunagawa S, Bork P. (2019). Disentangling the impact of environmental and phylogenetic constraints on prokaryotic strain diversity. bioRxiv 7. Domingo-Sananes MR, McInerney JO. (2019). Selection-based model of prokaryote pangenomes. bioRxiv Theme of the month: microbes to ecosystem functioningI wanted to get back to some of main goals in microbial ecology: relating microbial composition (or changes in composition) to function. This part of microbial ecology can lean heavily on the work done in community ecology from macroorganisms. In particular, there is a need to move beyond describing patterns of species richness and diversity to get at the functional consequences of this variation across spatiotemporal scales. This allows the use of a trait-based framework to really link traits to ecosystems and to link them inevitably to measures of diversity. As a primer, I would highly recommend reading this book chapter by Enquist et al (2013) which provides a thorough read through of trait based frameworks, including its influences from Grime's Mass Ratio and Metabolic Scaling Theory - both of which can be applied to microbial systems. Without further ado, here are some recent papers tackling this topic: Linking Microbial Diversity to FunctioningTraits underlie an organism's response to both biotic and abiotic factors. These traits will underline a particular organism's geographic distribution. For the most part, studies infer traits from microbial surveys, but linking these remain incredibly difficult. For one, it's impossible to quantify every trait for every microbes in a system. Of course, we can borrow from community ecology and concentrate on the more abundant, dominant members in a system (e.g., some of my work on Curtobacterium [1]). But this requires a lot of work, from isolation, characterization, developing relevant assays, collecting geographic distribution data, etc. Much more feasible is assaying for community-wide "traits" or determining function from aggregated functional responses (e.g., respiration). These approaches should allow for the formulation of hypotheses on microbial trait responses (for more see these two recent reviews here [2,3,4]). Environmental Gradients
Plants to soil to ecosystemsGoing to finish off with 3 papers I liked this past year or so. They are all related to soil microbiomes so sorry for any marine or human microbiome people out there! Do plants, bacteria, and fungi respond similarly by the same environmental variables? A recent study applies trait-based ecology to investigate the effects of temperature [8]. Specifically, should shifts in temperature correspond to shifts in plant traits and microbial function? And are there ecological feedbacks between plants and their microbes? Speaking of the soil microbiome affecting plant health. Wei et al. found that small variation in the initial soil microbiome can affect the health of plants throughout their experiment [9]. These results highlight the plant disease dynamics can be driven by highly deterministic processes based on the microbial composition and functional properties. Lastly, plants can regulate their rhizosphere microbial community by releasing chemical exudates from their roots [10]. The release of these exudates follows a "chemical succession" of microbes driving community assembly. Papers:1. Chase AB, Gomez-Lunar Z, Lopez AE, Li J, Allison SD, Martiny AC, Martiny JBH. (2018). Emergence of soil bacterial ecotypes along a climate gradient. Environmental Microbiology 20: 4112-4126.
2. Lajoie G, Kembel SW. (2019). Making the most of trait-based approaches for microbial ecology. Trends in Microbiology 27: 814-823. 3. Wang JT, Egidi E, Li J, Singh BK. (2019). Linking microbial diversity with ecosystem functioning through a trait framework. Journal of Biosciences 44: 109. 4. Malik AA, Martiny JBH, Brodie EL, Martiny AC, Treseder KK, Allison SD. (2019). Defining trait-based microbial strategies with consequences for soil carbon cycling under climate change. The ISME Journal 1-9. 5. McGill BJ, Enquist BJ, Weiher E, Westoby M. (2006). Rebuilding community ecology from functional traits. Trends in Ecology and Evolution 4: 178-185. 6. Glassman SI, Weihe C, Li J, Albright MBN, Looby CI, Martiny AC, Treseder KK, Allison SD, Martiny JBH. (2018). Decomposition responses to climate depend on microbial community composition. Proceedings to the National Academy of Sciences 115(47): 11994-11999. 7. Rath KM, Maheshwari A, Rousk J. Linking microbial community structure to trait distributions and functions using salinity as an environmental filter. mBio 10(4): e01607-19. 8. Buzzard V, Michaletz ST, Deng Y, He Z, Ning D, Shen L, Tu Q, Van Nostrand JD, Vooreckers JW, Wang J, Weiser MD, Kaspari M, Waide RB, Zhou J, Enquist BJ. (2019). Continental scale structuring of forest and soil diversity via functional traits. Nature Ecology and Evolution 3: 1298-1308. 9. Wei Z, Gu Y, Friman VP, Kowalchuk GA, Xu Y, Shen Q, Jousset A. (2019). Initial soil microbiome composition and functioning predetermine future plant health. Science Advances 5(9): eaaw0759. 10. Zhalnina K, Louie KB, Hao Z, Mansoori N, da Rocha UN, Shi S, Cho H, Karaoz U, Loque D, Bowen BP, Firestone MK, Northen TR, Brodie EL. (2018). Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nature Microbiology 3: 470-480. |
AuthorSome thoughts on some (small) things Archives
May 2023
Categories |
 RSS Feed
RSS Feed