|
Our new paper out in ISME investigates the importance of biotic interactions in structuring microbial communities at local spatial scales. Check out our "Behind the Paper" for more information!
0 Comments
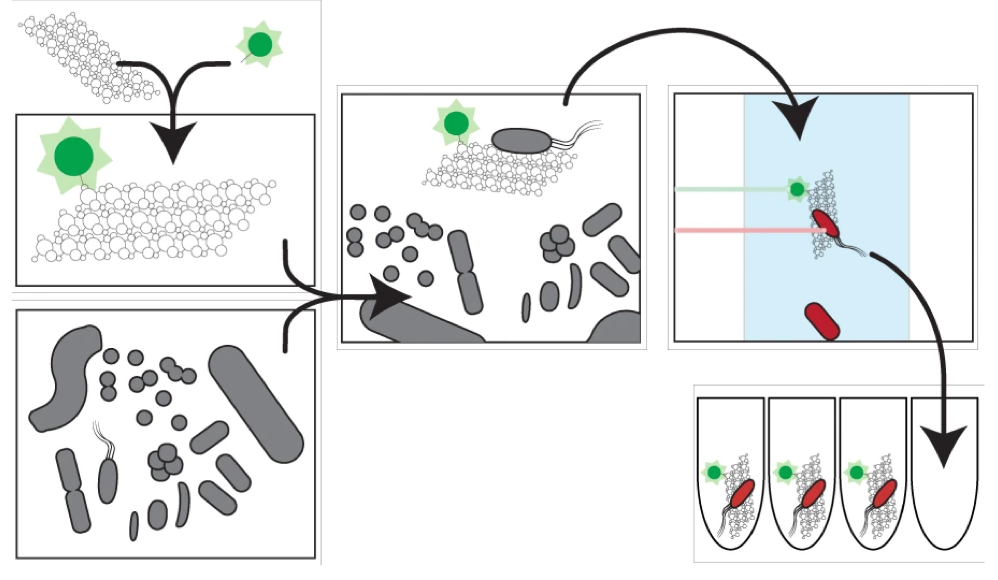
Been thinking a lot about metagenome assembled genomes (MAGs) and the potential for discovering novel microbial lineages. These offer a lot of insights into the metabolic function across diverse microbes with the potential for connecting previously undiscovered taxa to key biogeochemical processes. I figured I would condense my thoughts into this blog post in case it was of interest for any other microbial ecologists or microbiome scientists out there. Just my two cents on the topic... Brief Background on MAGsMost bacteria evade culturing efforts and most of the reference genomes in databases provide a limited, and highly biased, perspective of microbial diversity. With the advancements of sequencing technology and computational resources, it was becoming increasingly more apparent that creative and novel approaches could be used to assess microbial community composition. One of the earliest (if not the first?) approach using this "culture-independent" method was from Tyson et al. (2004) that looked at a relatively simple microbial community in biofilms [1]. Basically, we can sequence the entire genetic composition of the community, use genomic signatures of the abundant members, and cluster the contigs into similar "bins".
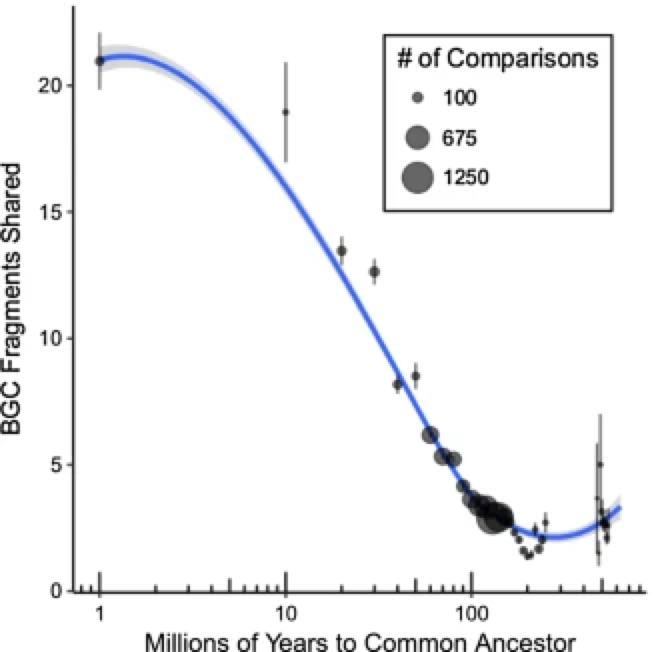
Modern Approaches and ApplicationsI can spend all year covering the recent papers using MAGs, but I will focus on a few of my favorites that are relatively recent. In general, most of the following papers are derived from more or less the same dataset [2]. Using dozens of metagenomes, the authors were able to reconstruct nearly 800 MAGs from a really complex and diverse soil community. By correlating environmental metadata with metabolic potential, the authors discovered differential metabolic pathways across MAGs that were directly associated with environmental gradients. In addition, this approach enabled insights into what the heck Acidobacteria might be doing. For those unfamiliar, this phyla of bacteria are incredibly difficult to culture, but are one of the most abundant phyla in soil systems. However, we barely know anything about them! The Acidobacteria MAGs encode a lot of CAZy enzymes, which are responsible for the breakdown of carbohydrates. Another earlier study also highlighted this phyla as a possible source for the discovery of new small molecules or specialized metabolites [3]. Furthermore, the distribution of the biosynthetic gene clusters (BGCs) encoding the production of these specialized metabolites varied across bacterial lineages and were correlated to environmental parameters, including soil depth and plant vegetation [4]. In any case, what I really like about these papers are the thorough work in characterizing these MAGs, especially the open source data and clear methodologies to complete such analyses. Just read the methods section in [2] and see what I mean - anyone can follow along and replicate the work done! Papers:1. GW Tyson, J Chapman, P Hugenholtz, EE Allen, RJ Ram, PM Richardson, VV Solovyev, EM Rubin, DS Rokhsar, JF Banfield. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature
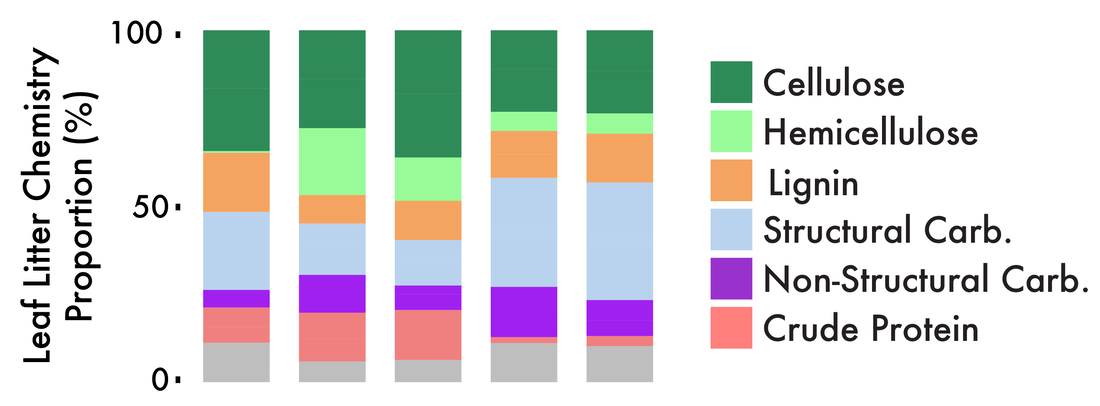
DOI: 10.1038/nature02340 2. S Diamond, PF Andeer, Z Li, A Crits-Christoph, D Burstein, K Anantharaman, KR Lane, BC Thomas, Chongle Pan, TR Northen, JF Banfield. (2019). Mediterranean grassland soil C–N compound turnover is dependent on rainfall and depth, and is mediated by genomically divergent microorganisms. Nature Microbiology DOI: 10.1038/s41564-019-0449-y 3. A Crits-Christoph, S Diamond, CN Butterfield, BC Thomas, JF Banfield. (2018). Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature DOI: 10.1038/s41586-018-0207-y 4. AM Sharrar, A Crits-Christoph, R Méheust, S Diamond, EP Starr, JF Banfield. (2020). Bacterial secondary metabolite biosynthetic potential in soil varies with phylum, depth, and vegetation type. mBio DOI: 10.1128/mBio.00416-20 For this week, I will be reading the paper out of the Buckley group entitled: "Competitive Exclusion and Metabolic Dependency among Microorganisms Structure the Cellulose Economy of an Agricultural Soil" in mBio I am pretty excited for this one as it kinda perfectly blends my passion for soil science (and C cycling) and the production of specialized metabolites (like antibiotics). Let's get this going! Cellulases and leaf litter decomposition
Bacteria that encode these cellulases are pretty well-distributed across the bacterial domain; however, many of the analyzed genomes lack complete degradation pathways for cellulose utilization [2]. While this is great to know the phylogenetic distribution of known cellulose degraders, a comparitive genomics perspective is limited as it cannot relate the genomic potential to community dynamics. This is where the field of carbon utilization is super amazing!
Physiological trade-offsNow that we have a brief background, let's dive in. Just a reminder that bacteria reside in communities. Meaning bacteria will have different life-history strategies like being a cellulose degrader. But if your neighbor is already releasing the enzymes to break down the cellulose, why would other bacteria need to also release MORE cellulases that require investment by the cell to produce and excrete. This means we can have a range of "cellulose utilizers" ranging from primary (and independent from other bacteria) degraders, incomplete cellulase degraders (only encoding some of the pathway), and opportunistic cheaters (who just gobble up the byproducts). This is where a major thought experiment can be underway and we can ask: Why invest in extracellular enzyme production if others are just going to cheat? In other words, how does the bacteria "guarantee" a return on its investment? Hint: antibiotics!!! Main resultsThe authors [4] use [13C]cellulose to group bacterial soils into a gradient of cellulose incorporation and used shotgun metagenomics to construct MAGs (metagenome assembled genomes). As the authors note, the MAGs were unable to recover abundant organisms and used a higher-level binning into "phylobins" at the Order level, as a side note they justify this by as the traits should be conserved at a phylogenetic level. Unsurprisingly, certain phylobins were enriched in [13C]cellulose while others were not. To get at the mechanisms driving these observations, the authors selected a representative genome (a bit unclear on this part - anyone want to help?) to compare the genomes for genomic traits, including genes related to surface attachment, motility, and specialized metabolism. [Funny result: the phylobin Cellulomonas was NOT enriched by [13C]cellulose.] The authors find that [13C]cellulose enriched phylobins produce more carbohydrate-active enzymes and more antibiotics, suggesting an ecological trade-off. One question here is: there is a general physiological trade-off in growth v yield. However, the authors note here that the [13C]cellulose-enriched taxa do both, grow faster AND make the investment in extracellular molecules, including both carbohydrate-active enzymes and specialized metabolites. Outstanding questions:1. How were the SM gene clusters linked to the proteomics? I am not familiar with proteomics at all, so generally curious how this was done. 2. What are the advantages of the slower growing, non-cellulolytic bacteria? If they cannot digest the substrate nor grow faster than the degraders, how do they persist in the community? Anyone have additional thoughts??? Papers:1. Chase AB, Gomez-Lunar Z, Lopez AE, Li J, Allison SD, Martiny AC, Martiny JBH. (2018). Emergence of soil bacterial ecotypes along a climate gradient. Environmental Microbiology
DOI: 10.1111/1462-2920.14405 2. Berlemont R, Martiny AC. (2012). Phylogenetic distribution of potential cellulases in bacteria. Applied and Environmental Microbiology DOI: 10.1128/AEM.03305-12 3. Doud DFR, Bowers RM, Schulz F, De Raad M, Deng K, Tarver A, Glasgow E, Meulen KV, Fox B, Deutsch S, Yoshikuni Y, Northen T, Hedlund BP, Singer SW, Ivanova N, Woyke T. (2020). Function-driven single-cell genomics uncovers cellulose-degrading bacteria from the rare biosphere. The ISME Journal DOI: 10.1038/s41396-019-0557-y 4. Wilhelm RC, Pepe-Ranney C, Weisenhorn P, Lipton M, Buckley DH. (2021). Competitive exclusion and metabolic dependency among microorganisms structure the cellulose economy of an agricultural soil. mBio DOI: 10.1128/mBio.03099-20 Hey everyone. I know it has been a long time since my last post, but I got completely consumed with work and adjusting to the new "norm" as everyone else has. I know this is not a unique situation for me, but definitely takes a lot out of someone. I am excited to resume some blogging and highlighting some recent papers I come across. I will try and keep these regularly updated, and if anyone has any requests or recommendations, please send them my way - it is much appreciated! In any case, one project, in particular, that has taken up a large portion of my time is looking at how specialized metabolism contributes to bacterial differentiation. From finishing my PhD a couple years ago, I wanted to integrate biotic interactions into microbial biogeography. Most of my work so far has focused on the abiotic factors contributing to environmental distributions. However, these are pretty broad measurements and observed across pretty distinct ecosystems. As a side note, we have some pretty exciting results coming soon describing evolutionary adaptation to abiotic factors as well for Curtobacterium in soils. Getting back to it. These biotic interactions are mediated through the production and secretion of specialized metabolites. These metabolite-mediated interactions are pretty well-documented in plants and animals, but they are pretty difficult to discern for microbes. Rather, these specialized metabolites have been almost exclusively viewed as sources for new natural products. While they have been instrumental for human health applications (i.e., pharmaceutical leads), microbes produce a wide variety of molecules with distinct biological properties mediating antagonistic interactions (i.e., antibiotics), resource acquisition (siderophores), or mutualistic interactions. As such, their abundance in nature and their implied ecological importance suggests these molecules should contribute to differentiation. My recent work is bridging the work from the natural product realm to integrate ecology and evolutionary theory as they relate to microbial diversification. And while I hope my work will be available to read soon (fingers crossed), I wanted to summarize some of the key papers and/or inspirational papers that contributed to my project. From biosynthetic genes to molecular innovation
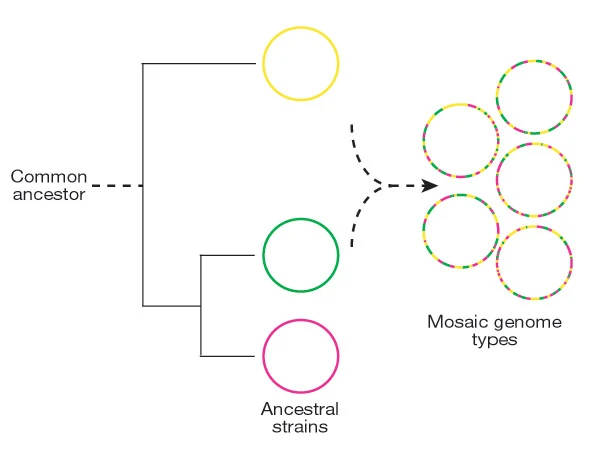
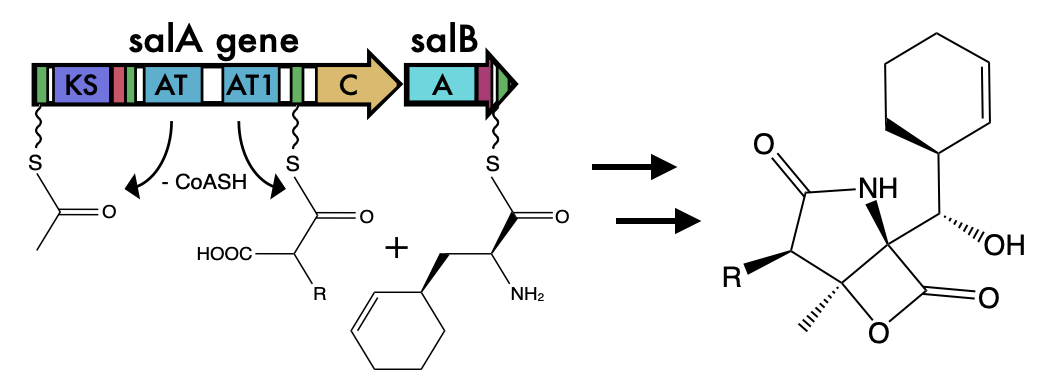
Examination of BGCs can reveal unique enzymology or biochemical interactions to prioritize BGCs for drug discovery. However, what is often ignored in the field of natural products, is how these BGCs evolve. This is of upmost importance because the evolutionary processes governing BGC diversity and distribution should contribute to molecular diversification and, possibly, affect their bioactivity. Within the last 10 years these questions have started to be asked in the field and found that horizontal gene transfer was a major contributor to BGC evolution. Surprisingly, these gene clusters, which can be quite large 50-90kbp, are thought to rapidly be gained and lost through HGT. The repeated observations for evidence of BGCs supports the ideas that these BGCs, and their cognate molecules, are integral to local adaptation. Here is a quote from a paper in 2016 (1): As a collective, the microbial genomes in each environment provide raw materials, the genomic building blocks that individuals can mix and match to create the BGCs that provide selective advantages. Adding ecological considerations to genetic and molecular studies allows the selection principles to be identified and fills out a molecule’s natural history, providing mechanistic insight into the manner in which BGCs are acquired and evolve. These reports were further supported in a systematic review of BGCs that concluded they have been "transferred horizontally at high frequency" (2). Certainly, BGCs can be acquired and result in drastic changes to an organisms' ecology and potentially allow for species transcending events. However, I was always intrigued by the extraordinary capability for these events to occur. The transfer of large, contiguous gene clusters across divergent organisms seems improbable, especially when the costs of HGT are considered ranging from integration of foreign DNA to the regulatory expression to the inherent risk of producing a toxic molecule (for more on HGT costs, definitely read this paper (3)). Despite all of this, the idea that HGT was a major contributor to BGC evolution persisted. After all, these BGCs must have come from somewhere, hence, they must have been acquired at some point in a lineage's evolutionary history. The identification of genomic islands in my third favorite Actino (everyone can probably guess who is #1), Salinispora, further provided a means for this mechanism (4). BGCs can evolve in a "plug and play" model for evolution to confer selective fitness advantages and be highly dependent on the local environment (5). Alternative models for BGC evolutionAt the same time, at least in Salinispora, there is evidence that the molecules are linked to species differentiation; for example, certain molecules are produced exclusively by a single species. This is akin to chemotaxonomy that is widely used in plants, and could also be a contributing factor in bacteria as well. This brings up quite the story. How can BGCs be frequently exchanged and we still observe species-specific signatures in molecule production. One way to look at this is to examine the distribution of BGCs among closely-related strains (stay tuned!!!). If HGT was THE major driver for BGC evolution, we would expect the distribution of BGCs to be random, that is strains that are more phylogenetically similar would have almost no correlation to BGC diversity. However, more and more recent evidence is coming out describing these BGCs (6) as conserved. So how can we reconcile these two seemingly contrasting models for BGC evolution? This is where the recent literature is super exciting, mainly a couple reviews from MG Chevrette (6,7). And the following quote summarizes some of these ideas: Rampant LGT of BGCs is a prevailing hypothesis in the field, but this view often overlooks the large evolutionary timescales in which an LGT event is observed. Vertical inheritance of BGCs represents an alternative mechanism. This is where I think the key is to resolving these ideas and can broken down into two main arguments: 1. HGT is not well-defined and vertical inheritance governs most of BGC diversity 2. BGCs can be readily modified through evolutionary processes to create diverse products
Second, BGCs can evolve de novo. Recent reviews have discussed these ideas and put forward that BGCs evolve neutrally, existing in generalist states to produce a diverse set of products (8). Once BGCs have evolved bioactivity, there are "endless modifications" of the ancestral pathways to increase potency and adapt to environmental conditions (9). So what are these modifications and what processes are governing BGC evolution? A lot of my ideas are presented in my upcoming manuscript that attempts to reconcile these observations to integrate evolutionary theory into natural products research. Briefly, rare HGT events occur and can contribute to large transcendent events. However, the vast majority of processes structuring BGC diversity and distribution are dictated by processes of vertical descent. At this level, fine-scale evolutionary processes (i.e., selection and recombination) can further drive BGC diversification with consequences for molecular innovation. Hope you enjoyed the read! Papers:1. Ruzzini AC & Clardy J. (2016). Gene flow and molecular innovation in bacteria. Current Biology.
DOI: 10.1016/j.cub.2016.08.004 2. Medema MH, Cimermancic P, Sali A, Takano E, Fischbach MA. (2014). A systematic computational analysis of biosynthetic gene cluster evolution: lessons for engineering biosynthesis. PLoS Computational Biology. DOI: 10.1371/journal.pcbi.1004016 3. Baltrus DA. (2013). Exploring the costs of horizontal gene transfer. Trends in Ecology and Evolution. DOI: 10.1016/j.tree.2013.04.002 4. Ziemert N, Lechner A, Wietz M, Millán-Aguiñaga N, Chavarria KL, Jensen PR. (2014). Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinispora. PNAS. DOI: 10.1073/pnas.1324161111 5. Letzel AC, Li J, Amos GCA, Millán‐Aguiñaga N, Ginigini J, Abdelmohsen UR, Gaudêncio SP, Ziemert N, Moore BS, Jensen PR. (2017). Genomic insights into specialized metabolism in the marine actinomycete Salinispora. Environmental Microbiology. DOI: 10.1111/1462-2920.13867 6. Chevrette MG, & Currie CR. (2019). Emerging evolutionary paradigms in antibiotic discovery. Journal of Industrial Microbiology & Biotechnology. DOI: 10.1007/s10295-018-2085-6 7. Chevrette MG, Gutiérrez-García K, Selem-Mojica N, Aguilar-Martínez C, Yañez-Olvera A, Ramos-Aboites HE, Hoskisson PA, Barona-Gómez F. (2020). Evolutionary dynamics of natural product biosynthesis in bacteria. Natural Products Reports. DOI: 10.1039/C9NP00048H 8. Noda-Garcia L & Tawfik DS. (2020). Enzyme evolution in natural products biosynthesis: target- or diversity-oriented? Current Opinion in Chemical Biology. DOI: 10.1016/j.cbpa.2020.05.011 9. Fewer DP & Metsä‐Ketelä M. (2020). A pharmaceutical model for the molecular evolution of microbial natural products. The FEBS Journal. DOI: 10.1111/febs.15129 Following my previous post, I will be covering some articles in the recent special issue of the Philosophical Transactions of the Royal Society B entitled "Conceptual challenges in microbial community ecology". I will try and expand on topics and connect outside work as well. This will not (at least hopefully) be a summary of the articles but my insights into the topics covering microbial ecology as a field. And if you want to read along, that previous post contains the schedule of the papers for each week. This week's highlight is "Linking regional shifts in microbial genome adaptation with surface ocean biogeochemistry" by CA Garcia, GI Hagstrom, AA Larkin, LJ Ustick, SA Levin, MW Lomas, and AC Martiny from the University of California, Irvine. Using 'omics to inform biogeochemical cyclesIn any environmental (or even host-associated) microbiome study, the typical ultimate goal is to relate the patterns, observations, and mechanisms driving microbial composition to ecosystem processes. This allows for predictions on how variation in abiotic factors (e.g., global climate change) will affect global biogeochemical cycles. This is not a trivial task as it requires understanding how the microbial community (and its relevant members) might change to the abiotic factors and how the resulting function will be affected (Figure). 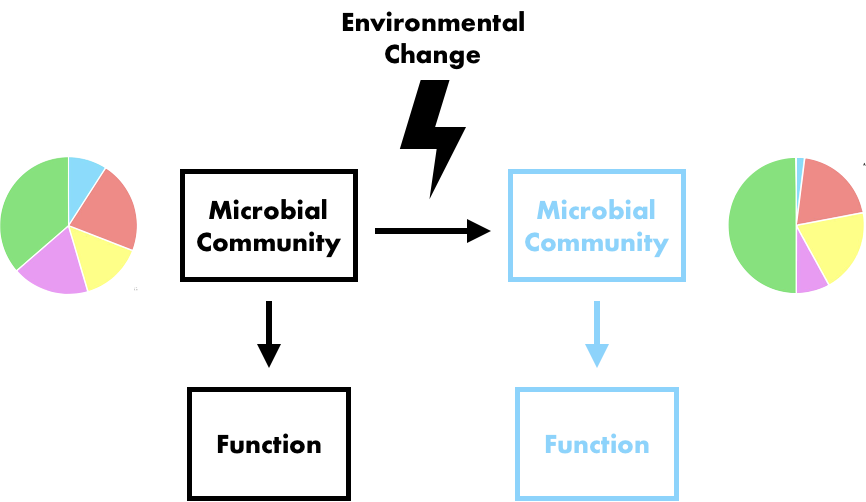 This framework is extremely powerful as it allows a predictive approach by considering the microbial traits themselves. A focus on microbial traits enables the ability to link community dynamics, environmental responses, and ecosystem processes. However, this "trait-based framework" (as a side note, highly recommended reading [1]) is often difficult to assess because it requires knowing which taxa and/or traits are useful biomarkers contributing to ecosystem processes. In this week's paper by Garcia et al. [2], the authors examine the ratios of carbon:nitrogen:phosphorus (C:N:P) and how microbes contribute to these global carbon and nutrient cycles. Typically, nutrient quantifications in the environment are hard to discriminate; for example, how to tell the difference between N availability if it's in the form of ammonium or nitrate? Well, the authors in this paper took a different perspective and thought about using microbial traits (or the genes encoding these traits) to infer relative nutrient conditions. This is made possible by the assumption that these genes within certain key taxa (i.e., Synechococcus and Prochlorococcus) will reflect "rapid" adaptation to nutrient conditions. For instance, in N-stressed environments cells will encode for more varieties of N uptake (e.g., urea, nitrite, and nitrate assimilation). This works in this system as decades of work underlies the distribution of nutrient niche partitioning in these two sister cyanobacterial genera (for more information, you can read my previous post here about the evolution of N genes in Prochlorococcus [3]). Further, these photosynthetic microbes largely contribute to global carbon cycles. A great question becomes, in other systems, which taxa and/or which genes would be adequate biomarkers to assess to link to ecosystem processes? Biogeography of nutrient-related genesThis paper utilized a global sampling scheme across multiple oceans, latitudes, and used various technical characterizations, such as metagenomics, flow cytometry, particulate organic matter, nutrient measurements, etc. The crux of this paper hangs on reliable classification of nutrient-related genes to nitrogen, phosphorus, and iron and their taxonomic origin. Again, this really only becomes convincing due to the decades of work into this system. Using a curated reference database of the Pro and Syn nutrient-genes, the authors could mine out reads from the metagenomic data to assess the diversity and distribution of these genes across various oceanic stations. After normalizing for single-copy marker genes, the authors found that certain oceans were more or less enriched in specific nutrient genes. These gene abundances corresponded to the environmental conditions, suggesting that the presence/absence of nutrient genes are directly related to adaptation to environmental conditions (Figure). 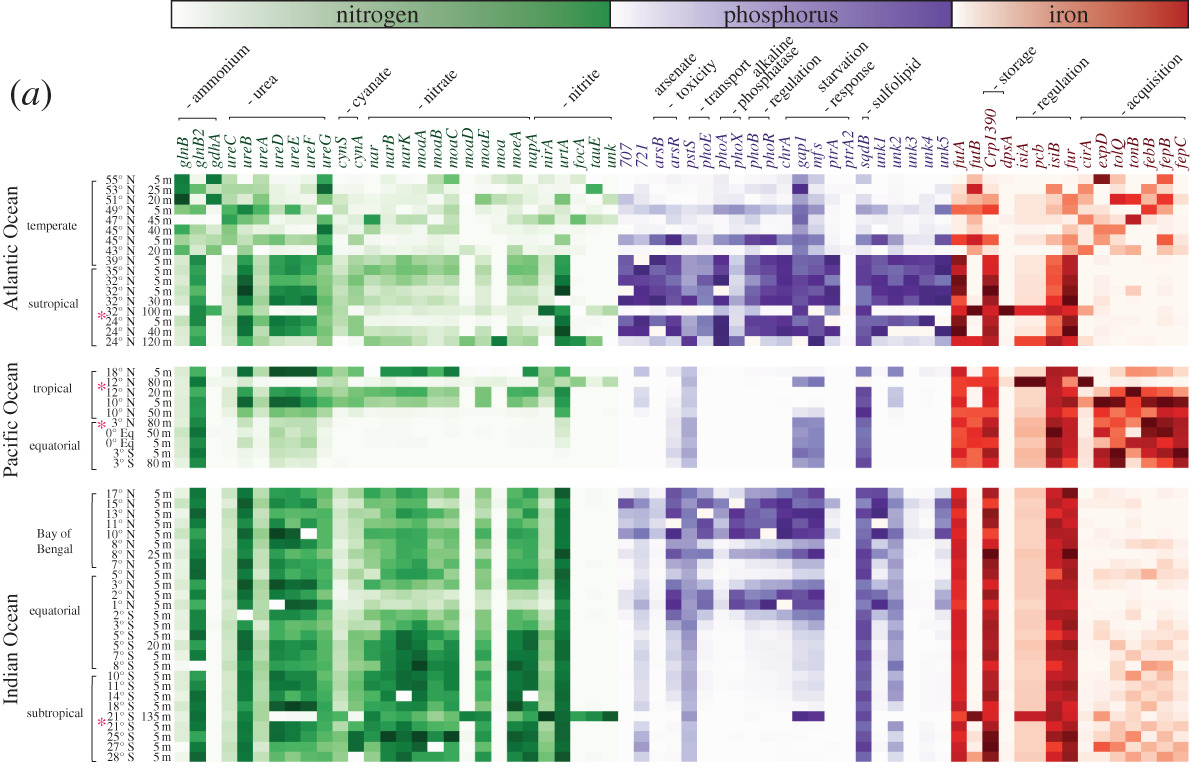 Variation among nutrient-related gene frequencies between sampling stations for Prochlorococcus Further, the authors found that these nutrient gene indices correlated to the measured inorganic nutrient concentrations and uptake; whereas, N gene coverage decreased with increased N uptake. Together, the inclusion of the nutrient gene indices in the trait-based models improved the predictions of carbon : phosphorus. By moving beyond strictly abiotic measurements, and using the microbiome, the authors could significantly improve predicted shifts in the elemental stoichiometry of marine communities. Papers: 1. Enquist BJ, Norberg J, Bonser SP, Violle C, Webb CT, Henderson A, Sloat LL, Savage VM. (2015). Scaling from traits to ecosystems: developing a general trait driver theory via integrating trait-based and metabolic scaling theories. Advances in Ecological Research.
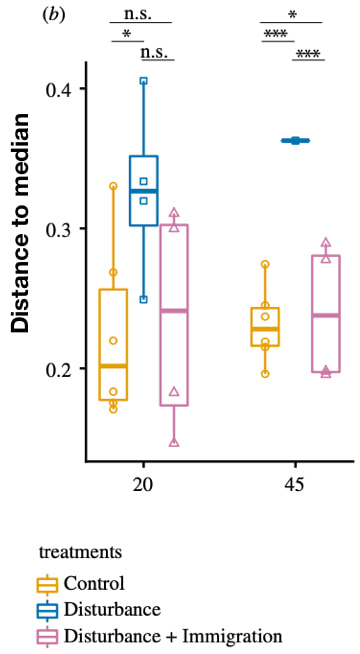
DOI: 10.1016/bs.aecr.2015.02.001 2. CA Garcia, GI Hagstrom, AA Larkin, LJ Ustick, SA Levin, MW Lomas, AC Martiny. (2020). Linking regional shifts in microbial genome adaptation with surface ocean biogeochemistry. Phils Trans. R. Soc. B. DOI: 10.1098/rstb.2019.0254 3. Berube PM, Rasmussen A, Braakman R, Stepanauskas R, Chisholm SW. (2019). Emergence of trait variability through the lens of nitrogen assimilation in Prochlorococcus. eLife DOI: 10.7554/eLife.41043 Following my previous post, I will be covering some articles in the recent special issue of the Philosophical Transactions of the Royal Society B entitled "Conceptual challenges in microbial community ecology". I will try and expand on topics and connect outside work as well. This will not (at least hopefully) be a summary of the articles but my insights into the topics covering microbial ecology as a field. And if you want to read along, that previous post contains the schedule of the papers for each week. This week's highlight is "Dormancy dynamics and dispersal contribute to soil microbiome resistance" by JW Sorensen & A Shade from Michigan State University. Microbial composition and disturbancesAll communities are constantly subjected to some type of disturbances. We learn about the gravity of these disturbances in "Intro to Ecology" courses with the famous intermediate disturbance hypothesis (IDH), popularized by JH Connell. Essentially, a disturbance occurs killing/removing individuals from the community and, thus, freeing up resources. The IDH predicts that at low disturbances, species diversity is decreased due to increased competition, leading to competitive exclusion. High disturbances, species turnover is greatest. Thus, intermediate disturbances would provide the highest species diversity in a given community. Allison & Martiny proposed that this idea of disturbances could impact microbial communities as well; in particular, that the community composition could shift or not depending on the system's ability to either be resistant (no compositional changes), resilient (return to original composition), functionally redundant (composition change, but no functional change), or altered composition and function [1]. As a side note, Ashley Shade presented this article at a CMI conference in San Diego earlier this year referring to the Allison & Martiny paper as a "classic". This framework is the foundation for the article by Sorensen and Shade [2], Specifically, they wished to address the mechanisms by which microbial communities could respond to a disturbance by looking at its capacity to be resistant or resilient to change. Alternatively, with high disturbances would secondary succession lead to reassembly or recovery of the original community or would alternative stable states be possible. The authors concentrated on two mechanisms that could contribute to microbiome stability: dormancy and dispersal. Dormancy and Dispersal and FIREDormancy in bacteria is a widespread and can create an equivalent of a seed bank in microbiomes [3]. Essentially, dormant cells resuscitate to help recover local populations when resources become suitable again. Dispersal can also aid in recovery if successful immigrants can establish and support resistance and restore community composition in sensitive communities. To test these ideas, the authors utilize a super cool field site as the initial microbial community from the Centralia, a series of coal mine, underground fires that is ongoing from 1962. The underground fires constantly spreads, warming the surface soils to variable thermal conditions. Previous work by this group showed variability among recovered soil communities that they hypothesized was from dormancy dynamics. Using a mesocosm setup, initial microbial communities were collected from temperate soils in Centralia and split into 15 samples at 14C. After 4 weeks, 9 samples were subjected to thermal stress at 60C for 8 weeks (plus 2 more weeks for steadily increasing/decreasing temp.) before returning to 14C. Of those 9 samples, 4 of the thermal stressed communities received an inoculation from the control communities, simulating a dispersal event.
ConclusionsDispersal is a known mechanism contributing to community dynamics; however, it is often difficult to discern in microbial communities without careful experimental design [4]. Here. the authors used a highly-controlled system to examine the effects of dormancy and dispersal to a disturbance. Although dormancy was less pronounced, the authors deduce that resuscitation of thermotolerant members of the community contributed to the transitional phase of the thermal stressors by assaying the active community. Personally, I could use some more evidence here and how dormancy would pertain to community stability during this disturbance period. Further, the results in this setup, although needed to tease a part these mechanisms, make me wonder how you could extrapolate this to the natural community at Centralia. Papers:1. Allison SD & Martiny JBH. (2008). Resistance, resilience, and redundancy in microbial communities. PNAS.
DOI: 10.1073/pnas.0801925105 2. Sorensen JW & Shade A. (2020). Dormancy dynamics and dispersal contribute to soil microbiome resilience. Phils Trans. R. Soc. B. DOI: 10.1098/rstb.2019.0255 3. Lennon JT & Jones SE. (2011). Microbial seed banks: the ecological and evolutionary implications of dormancy. Nature Reviews Microbiology. DOI: 10.1038/nrmicro2504 4. Albright MBN, Chase AB, Martiny JBH. (2019). Experimental evidence that stochasticity contributes to bacterial composition and functioning in a decomposer community. mBio. DOI: 10.1128/mBio.00568-19 Following my previous post, I will be covering some articles in the recent special issue of the Philosophical Transactions of the Royal Society B entitled "Conceptual challenges in microbial community ecology". I will try and expand on topics and connect outside work as well. This will not (at least hopefully) be a summary of the articles but my insights into the topics covering microbial ecology as a field. This week's highlight is "Ecological and evolutionary mechanisms underlying patterns of phylosymbiosis in host-associated microbial communities" by KD Kohl from the University of Pittsburgh. This week's discussion was led by Sarai Finks, a PhD candidate in the Martiny Lab at UCI. She was nice enough to send me her notes. And if you are interested in anything related to horizontal gene transfer, plasmids, and how all this contributes to niche differentiation, give her a follow! Sarai Finks: [Twitter link] PhylosymbiosisThis review paper outlines why researchers should consider the intimate connection of host-microbe interactions, as they provide constant feedbacks affecting functional components of the host and, in turn, the microbiome. Therefore, to further our understanding of the ecology and evolution of host-microbe interactions, we should deeply consider the processes that contribute to the observed signal. In particular, a major driver in structuring host-microbe interactions is shared evolutionary history. The idea of phylosymbiosis, or as the author states: congruence between the evolutionary history of various host species and the community structures of their associated microbiomes Within host-microbe interactions, it is largely assumed that selection of the microbiome is largely driven by the host, filtering the pool of microbial colonizers to establish symbionts. This paper, specifically, wants to highlight other, less-studied processes that could contribute to community structure: dispersal, selection, drift, and diversification (Figure). 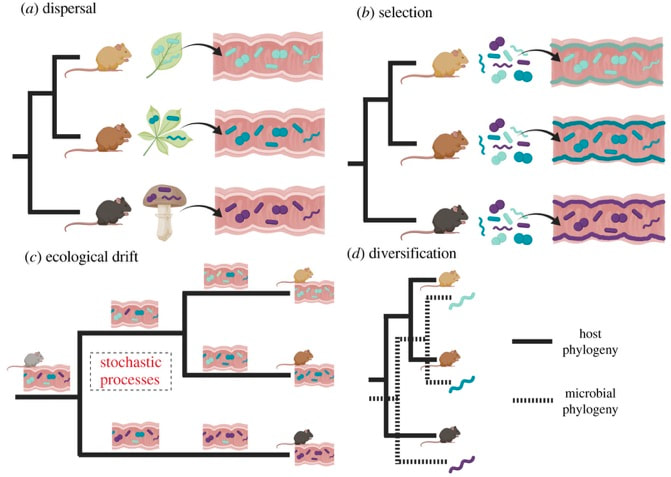 DispersalThe movement of microbes between environments can contribute to community structure, especially early on in colonization having profound priority effects. The author notes that host genetics could play a role, but dispersal could underlie a lot of observed phylosymbiotic patterns. The first paper I thought of reading this section was one of the more interesting attempts to disentangle these processes [2]. Using wild-type zebrafish and immune-deficient knockouts, this paper found that interhost dispersal could overwhelm the host genotype, largely homogenizing microbiomes across host genotypes. The idea that dispersal can play an important role in community assembly has been extensively studied in ecology (e.g., metacommunity ecology) but its role in host-associated microbiomes is definitely something I would like to see more! Selection
Ecological DriftDrift is an interesting topic with regards to host-associated microbiomes since it must be really hard to measure. With all the other confounding factors (i.e., host genotype, selection, dispersal) how can you tease these processes a part to assign variation due to ecological drift? I guess this would require extreme longitudinal sampling and possibly implementation of a reciprocal transplant in germ-free systems? DiversificationThe idea of diversification is an interesting, yet complex issue for host-associated microbiomes. For one, it is difficult to distinguish from co-phylogenies, or whether they represent co-speciation, a process whereby a symbiont speciates at the same time as another species. Further, these patterns can become even more convoluted from vertical and/or horizontal transmission of the microbiome from generation to generation. In sum, disentangling the relative importance of diversification in complex and diverse communities, and whether co-phylogenies represent co-speciation or host-shift speciation, remains poorly understood. Papers:1. Kohl KK. (2020). Ecological and evolutionary mechanisms underlying patterns of phylosymbiosis in host-associated microbial communities. Phils Trans. R. Soc. B.
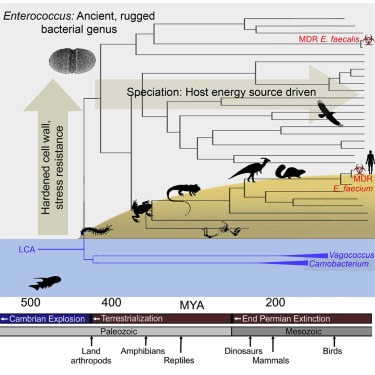
DOI: 10.1098/rstb.2019.0255 2. Burns AR, Miller E, Agarwal M, Rolig AS, Milligan-Myhre K, Seredick S, Guilemin K, Bohannan BJM. Interhost dispersal alters microbiome assembly and can overwhelm host innate immunity in an experimental zebrafish model. PNAS. DOI: 10.1073/pnas.1702511114 3. Lebreton F, Manson AL, Saavedra JT, Straub TJ, Earl AM, Gilmore MS. Tracing the Enterococci from Paleozoic Origins to the hospital. Cell. DOI: 10.1016/j.cell.2017.04.027 Following my previous post, I will be covering some articles in the recent special issue of the Philosophical Transactions of the Royal Society B entitled "Conceptual challenges in microbial community ecology". I will try and expand on topics and connect outside work as well. This will not (at least hopefully) be a summary of the articles but my insights into the topics covering microbial ecology as a field. This week's highlight is "Putting science back into microbial ecology: a question of approach" by JI Prosser from the University of Aberdeen. Getting from description to inferenceFor the first paper, we are covering the highly opinionated piece by James Prosser concerning the current state of microbial "ecology" [1]. This was actually something I was very excited to read since I have had a lot of ongoing discussions with colleagues concerning the exploding field of microbiome research. I have felt for a while, that a lot of papers coming out of the microbiome field are using the data to describe patterns, not pushing the boundaries of the mechanisms driving those patterns. This will be the theme for this article. Indeed, Prosser summarizes that out of 100 recent papers from AEM, EMI, FEMS, ISME, and Microbial Ecology 67 were descriptive and only 10 aimed to test a hypothesis - not great! The author begins by laying out what microbial ecology should be: understanding the relationships and interactions between microbes and their environment, whether it be host-associated, marine, soil, etc. However, as the author notes, that most studies fall under a spectrum of four approaches to address this ultimate goal. 1. Descriptive studiesThese are probably the most common of microbiome studies out there. Assessing the microbial community through a broad survey (typically of 16S rRNA reads) to catalogue who is there. These types of studies mimic the early ecology studies of natural history, describing general patterns of what organisms reside in what locations and habitats. Nowadays, these types of studies are relatively easy to do with private companies offering a one-stop-shop for microbiome workflows. This often leads to many studies plotting simple barplots or listing microbial taxa. As Prosser notes, these studies, due to their relative ease in generating, typically answer technical rather than scientific challenges as these studies lack aims or questions. And while our understanding of microbial diversity may expand with the discovery of new phylotypes (from 16S data) or MAGs (from shotgun data), they do not increase our understanding of microbial ecology. These datasets, as Prosser lays out, do not consider criteria for answering specific goals or aims related to how microbes interact with their environment. For instance, 16S surveys provide limited information - we cannot infer function, predict phenotype, or are unknown microbial taxa altogether. The same holds true for metagenomes, functional potential does not always translate (pun intended). Prosser admits not all descriptive studies ignore environmental parameters, but most still lack the direction of providing meaningful scientific results. Simply asking whether temperature influences microbial communities does not address the underlying mechanism of HOW temperature affects the microbial community. After reading this section, most graduate students around the world probably had a mild panic attack. If we cannot assess microbial diversity and correlate the composition to environmental parameters, what can we do? I find this first section a bit ambitious and harsh to the field. There are obvious advantages for these so-called descriptive studies. First and foremost, they provide a baseline to assess microbial community dynamics and how certain environmental parameters might affect community composition. Further, even with 16S rRNA (despite my over-harsh opinions on 16S myself...[2]) future hypotheses and questions can begin to be asked. Following our temperature experiment, having the knowledge that temperature is the main factor, we can ask how temperature can affect the physiology or metabolism of abundant members in this community. However, I do agree with Prosser that most studies do not follow through on these data-driven hypotheses. Far too often, 16S results are simply reported and over-extrapolated (e.g. "we saw an increase in this 1 OTU belonging to this order that has previously-known nitrogen fixers that we found in the literature"). These types of analyses are lazy and ignore the vast amounts of diversity within OTUs, taxonomic ranks, and ecological roles. Further, this does not attempt to progress anything in the field, a main point in Prosser's article. 2. Induction
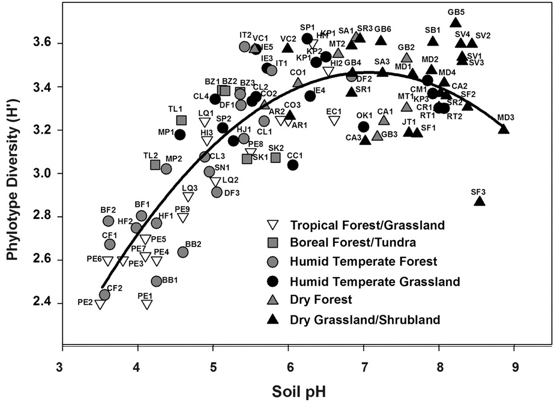
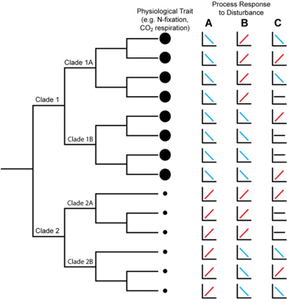
I know this seems trivial now, but at the time this was largely unknown. The idea of induction does not apply directly to this paper, in my opinion, but rather to the people who still view soil pH as a fundamental environmental parameter driving community composition. Soil pH is a widely all-encompassing parameters that could be influenced on the availability of nutrients, macro-fauna, abiotic parameters, etc. AND YET pH is still described as an important parameter. This is where I see Prosser's point. What are the mechanisms contributing to the pH differences that govern microbial community composition? 3. Inference to best explanationAfter reading this entire article, this is where I feel most of my work is on the border (or at least tries :P). I view the concepts of niche specialization and differentiation as my ultimate goal, by linking phylogeny and function and relating to environmental characteristics. As Prosser notes, microbiome studies, by their very name, should assume a relationship between phenotype of its members and the environment. However, broad-scale surveys of microbial communities lack the direct connection to physiological characteristics. In short, the correlations of taxa A in environment A does not actually provide causality. Prosser doubles down on this by also stating that most environmental parameters that are measured lack a priori consideration of the physiological differences between microbial taxa. This has been a topic brought up a few times by Prosser: the relationship between function and phylogeny. With the almost infinite amount of microbial diversity, how can we generalize community patterns or relate to phylogeny? Every trait cannot be accounted for and neither can every environmental parameters. However, not all traits and their relation to the environment are created equal. Some traits have a deep phylogenetic signal (Figure; also everyone go read this [4]). As another article within the issue will go over (stay tuned!), utilizing community data can reveal insights into environmental responses if the trait of interest is deeply conserved. The balance is assessing the appropriate functional trait and measuring the relevant, corresponding environmental parameters. Define the scientific aim, design the experiment, and test targeted hypotheses. In other words, don't fall into the blanket statements of pH controls soil microbial communities! 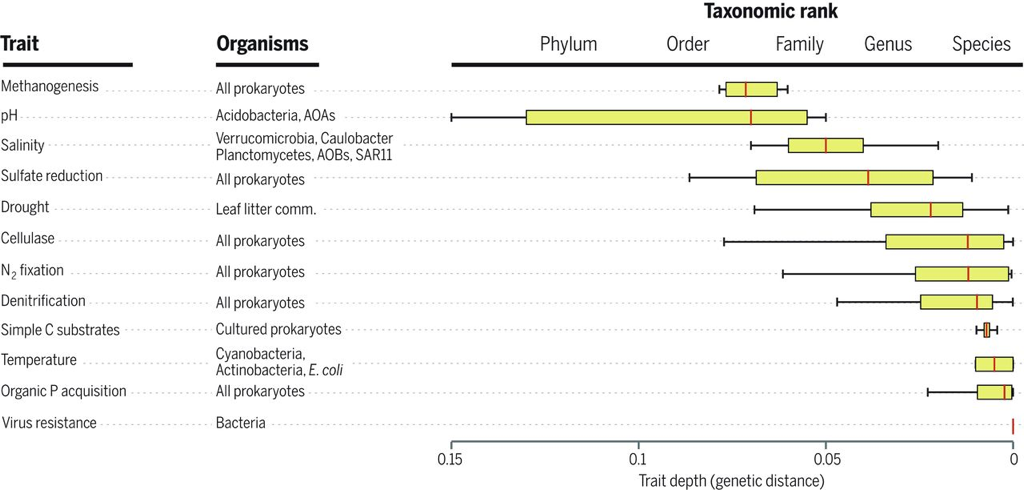 A box plot of the depth of clades within which taxa consistently share a trait measured as the genetic distance to the root node of a clade (bottom axis; usually of the 16S rRNA gene). From Martiny et al. Science. 2015. 4. Deduction and hypothesis testingProsser rounds out his spectrum of microbial ecology studies with deduction, Induction uses data to assess which hypothesis provides the best explanation. Deduction is the scientific method. This requires developing a "good hypothesis" - or bold, risky, meaningful! One issue is having targeted hypothesis being applied to the greater field of microbial ecology. As one person in the Microbial group at UCI so eloquently put it: "how do we make other people care about Curtobacterium?" What drives your science?That's all I got for this one - let me know your thoughts about the article, my analysis, suggestions, etc. So with that, I will leave you with this final quote from the article: The real limitation to our understanding of microbial ecology lies, not in a lack of techniques, but in a lack of motivation, enthusiasm, desire and courage to identify and ask significant scientific questions in advance of experimental work, and a lack of testable hypotheses and theory, i.e. lack of adoption of the basic scientific method. In this respect, it is worth considering, as a microbial ecologist, if you were to be given the answer to a single scientific question, or given a theory that explained a single phenomenon, what would be your question or phenomenon; in other words, what drives your science? Papers:1. Prosser JI. (2020). Putting science back into microbial ecology: a question of approach. Phils Trans. R. Soc. B.
DOI: 10.1098/rstb.2019.0240 2. Chase AB & Martiny JBHM. (2018). The importance of resolving biogeographic patterns of microbial microdiversity. Microbiology Australia DOI: 10.1071/MA18003 3. Fierer N & Jackson RB. (2006). The diversity and biogeography of soil bacterial communities. PNAS DOI: 10.1073/pnas.0507535103 4. Martiny JBHM, Jones SE, Lennon JT, Martiny AC. (2015). Microbiomes in light of traits: A phylogenetic perspective. Science DOI: 10.1126/science.aac9323 Conceptual challenges in microbial community ecologyWith all the pressing issues at hand both globally and locally for everyone, I have not felt the inspiration to make a blog post over the past few weeks. In light of that, I will just leave this recent issue of Philosophical Transactions of the Royal Society B - Biological Sciences. Just to give a primer of how amazing this issue is: Microorganisms play fundamental roles in environments from the human gut to the open ocean. Since technology now allows us to catalogue the enormous diversity of microorganisms in samples, a new challenge is to interpret these data by placing them into larger conceptual frameworks. This theme issue aims to highlight new advances and potential opportunities to do that. Four overarching themes emerge from the contributions: ScheduleUPDATE:
I was fortunate enough to get invited back to the UCI Microbial Group, who will be reading some articles from this issue throughout the Spring quarter (or the next 10 weeks). I will cover some articles on a weekly basis following their schedule and will update here for those who want to follow along. Week 1 - April 2nd "Putting science back into microbial ecology: a question of approach" JI Prosser DOI: doi/10.1098/rstb.2019.0240 Week 2 - April 9th "Ecological and evolutionary mechanisms underlying patterns of phylosymbiosis in host-associated microbial communities" KD Kohl DOI: doi/10.1098/rstb.2019.0251 Week 3 - April 16th "Dormancy dynamics and dispersal contribute to soil microbiome resilience" JW Sorenson & A Shade DOI: doi/10.1098/rstb.2019.0255 Week 4 - April 23rd "Experimental manipulation of selfish genetic elements links genes to microbial community function" SD Quistad, G Doulcier, PB Rainey DOI: doi/10.1098/rstb.2019.0681 Week 5 - April 30th "Linking regional shifts in microbial genome adaptation with surface ocean biogeochemistry" CA Garcia, GI Hagstrom, AA Larkin, LJ Ustick, SA Levin, MW Lomas, AC Martiny DOI: doi/10.1098/rstb.2019.0254 Week 6 - May 7th "Understanding the evolution of interspecies interactions in microbial communities" FA Gorter, M Manhart, M Ackermann DOI: doi/10.1098/rstb.2019.0256 Week 7 - May 14th "Phylogenetic conservation of soil bacterial responses to simulated global changes" K Isobe, NJ Bouskill, EL Brodie, EA Sudderth, JBH Martiny DOI: doi/10.1098/rstb.2019.0242 Week 8 - May 21st "How can microbial population genomics inform community ecology?" D VanInsberghe, P Arevalo, D Chien, MF Polz DOI: doi/10.1098/rstb.2019.0253 It has been a while since my last post. I needed to take some personal time and had to step away from science for a bit. After going through my first year at a postdoc, applying for faculty jobs (unsuccessfully), trying to finish up PhD projects, issues with intellectual and data sharing from colleagues, and some family emergencies along the way, I really needed to step back and focus on myself before I could come back to work. So apologies for the delay for those who follow these posts. I will be back on schedule from now on!
Framework to link microbial structure to function
Quantifying physiological traits What other endorsement do you need to read this next paper [3]. Understanding carbon use efficiency has wide-ranging applications from breaking down growth rate - yield tradeoffs to scaling CUE for ecosystem modeling. Using 23 diverse soil isolates, the authors investigated physiological traits, including growth rate on various substrates. By computing CUE metrics for each strain the authors could correlate these values to genomic predictions, such as rRNA operon copy number and extracellular enzymes. Some results included wide ranges in CUE temperature sensitivity across taxa, creating difficulties in predicting phylogenetic conservatism of an assumed complex trait. Brief note, I similarly found a wide range in carbon use across strains within a single genus and this variation was highly dependent on temperature [4] - what drives these differences?! Further, the authors found that genomic predicted traits did not correlate with CUE measurements, highlighting the difficulty in using genomic traits as reliable predictors. We always need to integrate physiology! Scaling from traits to response to whole communitiesThe best application of trait-based approaches in naturally occurring bacteria are probably the phytoplankton (specifically Prochlorococcus and Synechococcus). A new paper analyzed the distribution of picoeukaryotic phytoplankton with a global environmental abundance dataset combined with quantitative niche modeling [5]. The integration of the niche model enabled the authors to provide finer-taxonomic resolution to capture the high diversity of functional species within the phytoplankton groups (including differential responses to temperature and light). Together, the authors observed niche partitioning along a temperature gradient across the three major groups, which account for most of the photosynthetic biomass in tropical waters. Using metagenomics, microbial ecologists have gained an unprecedented insight into the functional potential of understudied taxa as well as whole communities. However, as the above paper suggests, the genomic potential doesn't always tell the entire story. This next paper utilizes time-resolved metatranscriptomics to understand how expressed genes in microbial communities contribute to functional observations [6]. By comparing metatranscriptomes across rhizosphere and bulk soil, the authors found a significant successional pattern across time. Further, functional genes indicated a rapid functional successional pattern as well with four major functional guilds emerging: specialization to root exudates, decaying roots, root biomass, and later stage roots. These overall patterns suggest that specific microbial taxa specialize on various spatiotemporal strategies related to both habitat (root v soil) and time. As a disclaimer to those interested in using metatranscriptomics, they are very messy datasets. Honestly, they make metagenomes look easy... Here, the authors capitalized on a well-known system (just check the author list!) that has previously created a unique reference database for this specific system, including single-cell genomes (SAGs), isolate genomes, and stable isotope probing metagenomes. Last paper I want to include really capture why assessing the (active) functional genes can better resolve community patterns. In particular, does relic DNA (essentially leftover DNA from dead organisms) mask community patterns [7]? Using fine-scale spatial sampling combined with temporal sampling, the authors demonstrate that, unsurprisingly, spatial variation largely explains community patterns. This is not new as distance decay relationships were an early observation for the argument FOR microbial biogeography patterns. What was interesting is the temporal dynamics. The observed temporal dynamics was hindered by the presence of relic DNA. By accounting for relic DNA in the samples, the authors found the temporal signal to be more evident. Papers:1. Bardgett RD, Caruso T. (2020). Soil microbial community responses to climate extremes: resistance, resilience and transitions to alternative states. Phil. Trans. R. Soc. B
DOI: 10.1098/rstb.2019.0112 2. Allison SD, Martiny JBH. (2008). Resistance, resilience, and redundancy in microbial communities. PNAS DOI: 10.1073/pnas.0801925105 3. Pold G, Domeignoz-Horta LA, Morrison EW, Frey SD, Sistla SA, DeAngelis KM. (2020). Carbon use efficiency and its temperature sensitivity covary in soil bacteria. mBio DOI: 10.1128/mBio.02293-19 4. Chase AB, Gomez-Lunar Z, Lopez AE, Li J, Allison SD, Martiny AC, Martiny JBH. (2018). Emergence of soil bacterial ecotypes along a climate gradient. Environmental Microbiology DOI: 10.1111/1462-2920.14405 5. Flombaum P, Wang WL, Primeau FW, Martiny AC. (2020). Global picophytoplankton niche partitioning predicts overall positive response to ocean warming. Nature Geoscience DOI: 10.1038/s41561-019-0524-2 6. Nuccio EE, Star E, Karaoz U, Brodie EL, Zhou J, Tringe SG, Malmstrom RR, Woyke T, Banfield JF, Firestone MK, Pett-Ridge J. (2020). Niche differentiation is spatially and temporally regulated in the rhizosphere. ISME DOI: 10.1038/s41396-019-0582-x 7. Carini P, Delgado-Baquerizo M, Hinckley ELS, Holland-Moritz H, Brewer TE, Rue G, Vanderburgh C, McKnight D, Fierer N. (2020). Effects of spatial variability and relic DNA removal on the detection of temporal dynamics in soil microbial communities. mBio DOI: 10.1128/mBio.02776-19 |
AuthorSome thoughts on some (small) things Archives
May 2023
Categories |

 RSS Feed
RSS Feed