Why do microbiologists have to invent new terms?!
A recent tweet (see below) brought up a great point and is something I have been thinking about for some time now. With all the new capabilities to assess the HUGE amounts of microbial diversity, I think it is time for some normalization of how we describe and discuss microbes. Just like any systematics in any organism, there are issues. However, the ability to describe evolutionary relationships has been at the foundation of evolutionary biology since Darwin and is integral to how we compare and assess various eco/evo processes affecting biodiversity and biogeography.
At some level, it's easy to differentiate species. For instance, I think everyone can tell the difference between a red algae and a horse. With microorganisms, it isn't as easily to differentiate individuals based on morphological attributes; and instead we lean heavier on genetic and molecular techniques. Commonly, microbiologists use conserved marker genes, such as the 16S rRNA gene, to classify and assess phylogenetic relatedness among isolates and/or culture-indepedent amplicon sequences. This has led to unprecedented progress in our understanding of microbial diversity and the underlying processes at relatively broad genetic resolutions in microbial communities. But just as easily as we can go out and assay thousands of microbial taxa in soil, we can just as easily over-interpret our findings. For instance, there is a desire to prematurely relate microbial studies to the enormous theoretical and empirical work done with macroorganisms. But, microbial ecology is in its infancy and, as such, I find the use of microbe-specific terminology informative and necessary.
OTUs, microdiversity, populations, and species, OH MY!
Now before I start my rant into microbial terminology, I want to point out that macroorganisms have their own confusing terminology (e.g., cryptic species). Ambiguous terminology is always something that will be an issue, but, nevertheless, remains an important topic when analyzing biodiversity patterns. In any case, specifically for microbial ecology and evolution, I think the fundamental issue arises from the inability to relate molecular methods to biological questions. Just as in any study, the molecular methods you choose must reflect your biological question because, as this commentary so nicely puts it, "in nature, there is only diversity" [1]. If we just go out and blindly sample microbial diversity, we cannot address the mechanisms contributing to observed biogeographical patterns.
Microbial species
The fundamental unit of biology is the species. For decades, microbiologists have tried to quantify what constitutes a species and when other clades should be reclassified. For this post, I will only concentrate on the newer techniques used for delineating microbial taxa.
Obviously this metric only examines genomic relatedness and does not consider whether environments or ecology contribute to species designations. I will come back to these ideas later, but for now let's move on to OTUs, or operational taxonomic units.
OTUs or ESVs cannot represent species
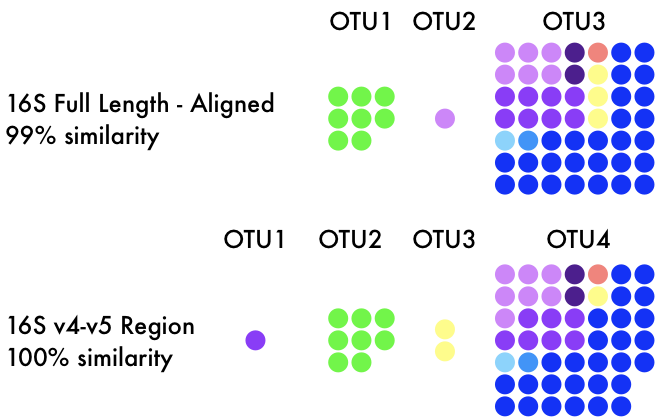
Increasingly, researchers are using culture-independent methods to assess previously unknown microbial diversity. The most popular of these methods is amplicon-based metagenomic analyses utilizing the 16S rRNA marker gene. With newer and newer technologies and computational resources, we can easily sequences hundreds of thousands of microorganisms from a given environmental sample. This has led to the implementation of delineating microbial taxa into operational taxonomic units (OTUs), but it is unclear how these taxonomic delineations represent more traditionally-defined terminology. Initially, some tried to relate OTUs to microbial species but these proved far too broad to quantify. For instance, a 3% divergence in the 16S rRNA gene (the most common threshold for OTU clustering) represents the same evolutionary divergence time as the origin of modern birds or roughly 150M years. Reread that last sentence and let it sink in...
With newer sequencing technologies came the reduction of sequencing breadth of the 16S rRNA gene. Most studies nowadays target a hypervariable region of the 16S rRNA gene (usually the V4/V5 region), which only targets a small section of the ~1600bp 16S gene. This is advantageous as we can ignore conserved, non-informative regions and sequence more individuals within the community. This has also led to a movement away from OTUs to exact sequence variants (ESVs) to quantify microbial diversity. This would allow each variant (or SNP) in the 16S gene to infer evolutionary history, providing a far more realistic depiction of microbial taxonomic units. Now this is where we circle back around again. Repeatedly, I have seen papers inferring "population dynamics" or "intraspecific variation" across microbial taxa by assessing differential distributions of ESVs (sound familiar? remember the birds...). I do not want to list papers, but I fear this is becoming more widely abused in practice as more and more microbial communities are assessed. But just as full genomes do not consider ecological factors, neither ESVs nor OTUs provide the resolution to infer functional traits or how those correspond to environmental factors. And I am being generous, there are examples of a single SNP in the entire genome contributing to diversification. Microdiversity
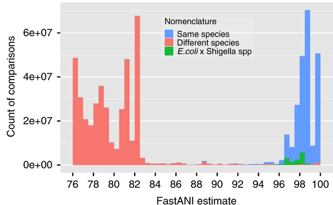
This is where the term microdiversity can be beneficial. I will define microdiversity as closely-related strains within the same taxonomic group, however defined. For ease of this argument, I will focus on the microdiversity within OTUs or ESVs (hereafter referred to just OTUs since its just a matter of the clustering threshold). As stated above, we know little of the variation within OTUs and how trait variability contributes to the distribution of microbial taxa. Moreover, we do not know how to delineate microbial species and sometimes even genera (see Escherichia and Shigella). Therefore, relating community analyses with corresponding OTU distributions is almost impossible to equate to frameworks devised for macroorganisms at the species level.
Moving towards a bacterial species definition - ecotype model
Incorporating genomic and ecological information (i.e., environmental distributions and phenotypes) allows a more robust picture to emerge for delineating microbial taxa. Cohan proposed adapting an "ecotype model" to delineate ecological populations, as he describes it [5]:
"ecologically distinct lineages (based on sequence analysis) and as a prognosis of future coexistence (based on ecological differences), are the fundamental units of bacterial ecology and evolution"
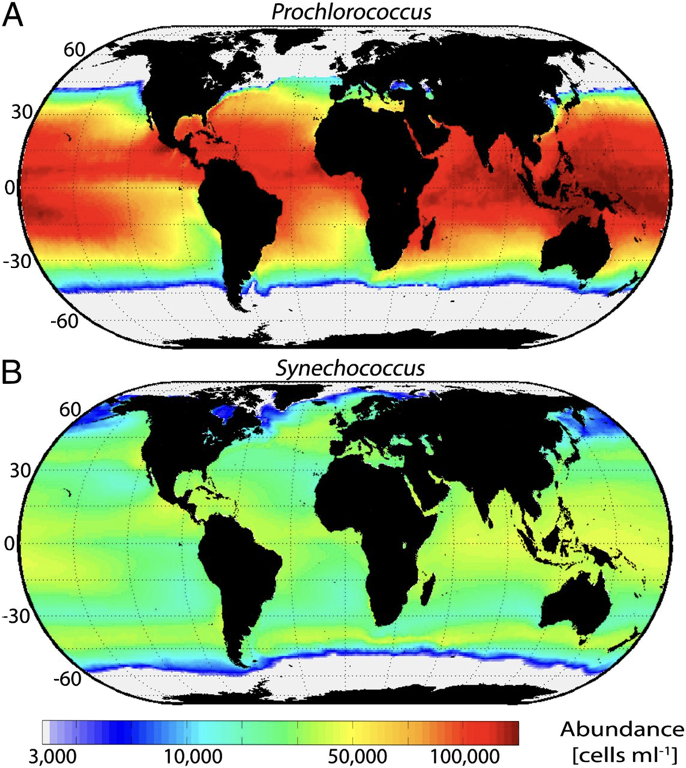
Essentially, he is describing the fundamental unit of biology, species (I know, ecotype means ecological population but I am equating it to species - can it get more confusing!). However, to get to this point requires extensive work in a microbe: collate genomic, phenotypic, environmental data, etc. to delineate ecotypes. And even if you can get to this point, there is still ambiguity to what level qualifies an ecotype as it can be a sliding scale. For instance, the heavily studied marine cyanobacterium, Prochlorococcus has been designated into several ecotypes, each corresponding to phenotypic traits with these traits highly correlated to environmental distributions (see other blog post here). Even in the best-known case of Prochlorococcus, we are still clustering genomes into ecotypes that diverge far below suggested ANI species boundaries (e.g., Pro ecotype HLII includes genomes <88% ANI).
Simply put, there is still A LOT of work to do. Evolutionary Processes and Populations
Lastly, I just want to end with microbial populations. Ideally, this is the level microbiologists need to get if we wish to understand the evolutionary processes driving speciation. Populations, by definition, are groups of individuals within the same species. By "jumping the gun" and moving from environmental community-level studies to "intraspecific variation" using ESVs we are doing ourselves a disservice. We need to learn to walk before we can run. And this requires extensive work to first describe species and then ask questions into the underlying population dynamics driving microbial diversification.
Populations are also groups of interbreeding individuals, but as asexual organisms, microbial are harder to pinpoint. However, we can use recombination as a metric for reproduction to identify possible population clusters. A recent study from MIT attempts to do just that, by defining populations to illuminate ecologically-distinct populations (figure below) [6]. 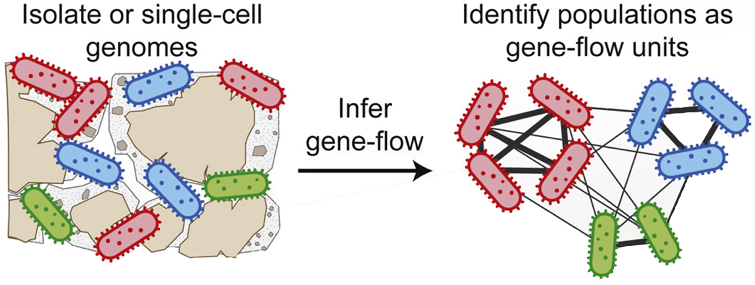
Papers
1. McLaren MR, Callahan BJ. (2018). In nature, there is only diversity. mBio 9: e02149-17.
2. Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. (2019). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nature Communications 9: 5114. 3. Chase AB, Karaoz U, Brodie EL, Gomez-Lunar Z, Martiny AC, Martiny JBHM. (2017). Microdiversity of an abundant terrestrial bacterium encompasses extensive variation in ecologically relevant traits. mBio 8: e01809-17. 4. Chase AB, Gomez-Lunar Z, Lopez AE, Li J, Allison SD, Martiny AC, Martiny JBHM. (2018). Emergence of soil bacterial ecotypes along a climate gradient. Environmental Microbiology 20: 4112–4126. 5. Cohan FM. (2006). Towards a conceptual and operational union of bacterial systematics, ecology, and evolution. Philos Trans R Soc Lond B Biol Sci. 361: 1985-1996. 6. Arevalo P, VanInsberghe D, Elsherbini J, Gore J, Polz MF. (2019). A reverse ecology approach based on a biological definition of microbial populations. Cell 178: 820-834.e14.
0 Comments
BackgroundWhy are there so many species and what are the limitations for the coexistence of N species? These are fundamental questions in ecology and evolution that have been at the foundation of these fields for centuries. On one hand, Hubbell's Neutral Theory [1] assumes that all species are essentially ecologically equivalent (does not actually claim this, just that neutral processes are greater than deterministic) and that community composition is dependent on stochastic processes (i.e., dispersal limitation and ecological drift) causing species abundances to vary. On the other side of the spectrum is niche theory, where species interactions (biotic) and environmental filtering (abiotic) drive community assembly and composition. For niche theory, Hutchinson (1951) and Tilman (1982) provide relatively nice explanations for species coexistence that can be summarized as species can coexist if they can partition their niche along some type of an environmental resource. In particular, the niche-based framework can be a POWERFUL approach because it can account for the links between evolution, (a)biotic environments, and the community [2]. This is because ecological niches are intricately linked to functional traits, as traits underlie an organism's response to abiotic and biotic conditions. Thus, we can examine the evolutionary history of functional traits to gain insights into niche diversification. Recently, these ideas have been explored in microbial ecology. For instance, my dissertation addressed how bacterial ecotypes have differential distributions due to variation in their functional traits [3]. More widely applicable reviews are also available exploring the phylogenetic conservatism of microbial traits [4]. However, we really are at the forefront of understanding the distribution of traits and how evolutionary processes structure microbial trait variability, especially among closely-related taxa. This is why I am really excited to discuss a recent publication concerning the "Emergence of trait variability through the lens of nitrogen assimilation in Procholorococcus" [5]. ProchlorococcusProchlorococcus (Pro) is a globally-distributed marine cyanobacterium that is a major contributor to global photosynthesis. Synechococcus (Syn) is a sister genera that is more abundant at higher latitudes, suggesting partitioning of environmental resources along temperature and nutrient gradients [6]. Within Pro, numerous work over decades in the Chisholm lab have uncovered fine-scale phylogenetic clades that correspond to physiological traits, such as pigmentation, growth rate, and nutrient utilization, and have subsequently been coined ecotypes. By sampling at various depths and across latitudes, Pro ecotypes exhibited differential geographic distributions that were highly correlated to environmental gradients, such as temperature and nitrate [7]. These distributions were corroborated by phenotypic trait assays (i.e., strain that grows better under higher temperatures were found in hotter geographic regions). Thus, by examining trait variation we can better understand the biogeographic distribution of microbes and its relation to niche partitioning.
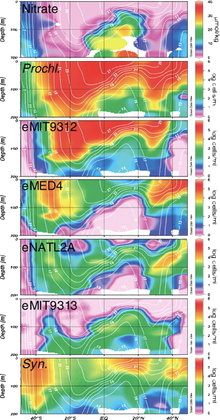
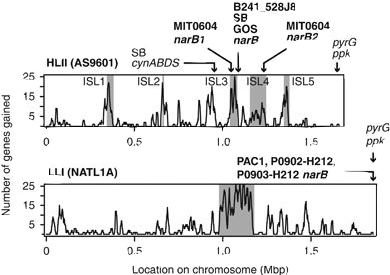
Patchy distribution of Nitrogen traits in ProBoth Pro and Syn are non-N fixing bacterium, and as N can be a limited nutrient for all phytoplankton, it represents a crucial environmental resource. All Syn can assimilate nitrate, but the genetic repertoire in Pro is only found in a few strains that are constrained to a couple ecotypes. This begs the question of whether genomic cluster containing the nitrate assimilation pathway was vertically inherited or if horizontal gene transfer (HGT) mediated the evolution of this trait.
To test this, the authors examined phylogenies for the upstream and downstream genes of the nitrate pathway. Almost universally, the phylogenies were highly congruent with the core genome phylogeny for each gene, rejecting evidence for high degrees of HGT. As further support, comparative genomics of the complete nitrate gene cluster across all Pro genomes revealed high synteny within clades, strengthening the argument for vertical inheritance. 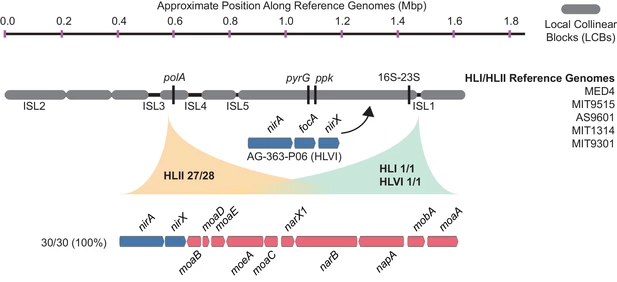 Location of the nitrate assimilation cluster in the high light clade (HL). All strains in the HL clade contain identical gene order and genomic location of the nitrate assimilation gene cluster. Homologous recombination shapes trait diversityTo recap, the patchy distribution of the nitrate assimilation gene cluster is not likely due to HGT. Another possibility is the cluster represents a defining trait within Pro clades and provides a differentiating trait within Pro. One such mechanism that reinforces this process is homologous recombination, as the rate of recombination is expected to exponentially decrease with increasing sequence divergence. When the authors investigated the relative role of homologous recombination in structuring the genetic diversity of Pro, they found it can represent a cohesive force shaping the genetic similarity within clades. Indeed, the high r/m (recombination to mutation ratio) suggests that recombination structures genetic diversity far more than mutation accumulation if the genes were diverging. Further, low nucleotide diversity of nitrate assimilation alleles suggests that gene-specific sweeps occurred most likely providing an advantageous trait delineating Pro clades. Evolution of N trait variabilityThe nitrate assimilation gene cluster has a complex evolutionary history that has been mediated through vertical inheritance and high rates of homologous recombination within Pro clades, driving stochastic gene gain/loss to lead to differentiation. However, one major anomaly exists: the absence of the nitrate gene cluster in basal Pro lineages. To address this, the authors finish with an evolutionary model describing the evolution of N and its relation to niche partitioning among Pro clades (see figure below). Briefly, competition and resource trade-offs facilitate partitioning of environmental resources (i.e., nitrate) that causes the stochastic loss of the nitrate gene cluster. When advantageous, homologous recombination has constrained divergence of the nitrate gene cluster even manifesting in gene-specific sweeps within clades. As clades further divergence, homologous recombination is depressed allowing for ecological differentiation. I just want to end with a quote from the paper to illustrate how difficult disentangling these evolutionary processes are; however, as I hope you can take away from this paper, how crucial insights into trait variability can elucidate both ecological and evolutionary processes. "Superficially, this emergent pattern in microbial trait variability might appear to be the result of horizontal gene transfer, but our evidence indicates that the observed patterns can be attributed to processes of vertical descent, gene loss, and recombination between close relatives that have operated throughout the entire radiation of Prochlorococcus." 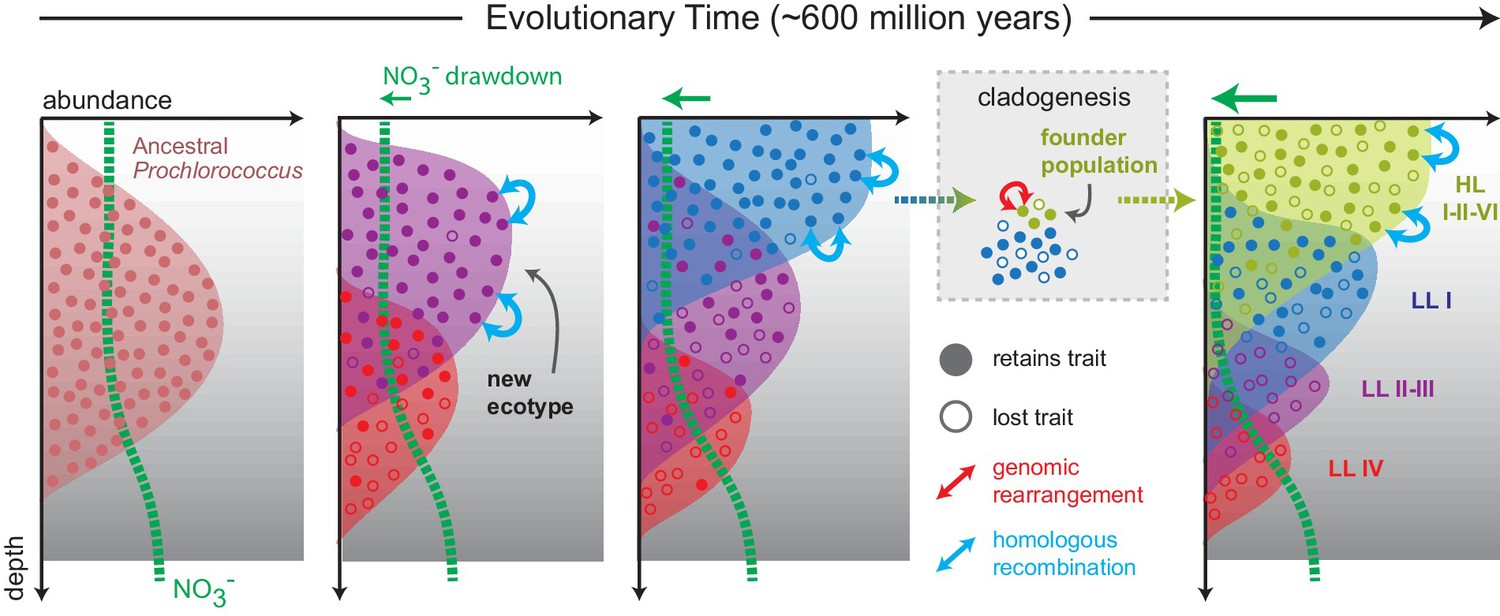 Papers1. Hubbell SP. (2001). The Unified Neutral Theory of Biodiversity and Biogeography. Princeton University Press.
2. Chase JM and Leibold MA. (2003). Ecological Niches. The University of Chicago Press. 3. Chase AB, Gomez-Lunar Z, Lopez AE, Li J, Allison SD, Martiny AC, Martiny JBHM. (2018). Emergence of soil bacterial ecotypes along a climate gradient. Environmental Microbiology 20: 4112–4126. 4. Martiny JBHM, Jones SE, Lennon JT, Martiny AC. (2015). Microbiomes in light of traits: a phylogenetic perspective. Science 350: aac9323. 5. Berube PM, Rasmussen A, Braakman R, Stepanauskas R, Chisholm SW. (2019). Emergence of trait variability through the lens of nitrogen assimilation in Prochlorococcus. eLife 8: e41043. 6. Flombaum P, Gallegos JL, Gordillo RA, Rincón J, Zabala LL, Jiao N, Karl DM, Li WKW, Lomas MW, Veneziano D, Vera CS, Vrugt JA, Martiny AC. (2013). Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proceedings to the National Academy of Sciences 110: 9924-9829. 7. Johnson ZI, Zinser ER, Coe A, McNulty NP, Woodward EMS, Chisholm SW. (2006). Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science 311: 1737-1740. 8. Berube PM, Biller SJ, Kent AG, Berta-Thompson JW, Roggensack SE, Roache-Johnson KH, Ackerman M, Moore LR, Meisel JD, Sher D, Thompson LR, Campbell L, Martiny AC, Chisholm SW. (2015). Physiology and evolution of nitrate acquisition in Prochlorococcus. The ISME Journal 9: 1195-1207. BackgroundA major goal in evolutionary biology is to understand the origin and maintenance of genetic variation and diversity. This framework allows us to identify the processes governing species distributions and diversity. In microbes, it is difficult to delineate species boundaries and identify populations undergoing diversification. Most studies employ comparative genomics by collating closely-related genomes from various databases. However, these analyses do not consider whether strains were from the same habitat and, thus, we cannot examine whether they are a part of the same populations of interacting genotypes. Being able to compare diverging populations allows us to identify the mechanisms contributing to microbial speciation and diversification. In theory, speciation is dependent on 1) selection leading to ecological differentiation and 2) barriers to recombination. This is similar to sexual organisms; however, the lack of sexual reproduction in microbes makes it harder to differentiate. These are all topics that fascinate me in microbial ecology and evolution. To this day (for a variety of reasons I will try and cover in another post), we still grapple with the mechanisms driving microbial diversification and speciation. The case of Vibrio provides an excellent story addressing these topics and was one of the more interesting studies that motivated me to pursue population dynamics in environmental microbes. Vibrio - a test case
Organic particles in the ocean are thought to be hotspots for community aggregation. Insomuch, that marine particle-attached communities can undergo ecological succession, where species turnover is dictated by resource availability. In this case, motile bacteria arrive first and begin the degradation of particles and later followed by secondary consumers of the carbon byproducts [2]. Population GenomicsFollowing the observations that Vibrio strains appeared to have preferences for particle size, a study lead by Shapiro sought to identify the genomic mechanisms of divergence potentially leading to bacterial speciation [3]. Utilizing 20 genomes from large (L) or small (S) particles sizes (proxy for habitat), they first found evidence for differentiation among closely-related strains. For perspective, the strains all shared identical 16S rRNA genes and >99% average amino acid identity across the genome. The relevant differences among these strains resided in 725 "ecoSNPs" that cluster in genomic space in 11 concentrated regions (see below figure). These ecoSNPs supported habitat divergence, while the rest of the genomic SNPs provide incongruence with the ecological split into distinct habitats. Further, these SNPs contain low within-habitat diversity suggesting these regions are a result from recent recombination and have subsequently swept through the populations, coined gene-specific sweeps. Analyses of the core and flexible genome support these conclusions as they identified core regions more frequently recombined within-habitat than between-habitat.  A) core genome phylogeny for chromosome I and II B) genome regions showing localization of ecoSNPs Ecological DifferentiationEvidence for high recombination within-habitat strains led the authors to inquiry how these populations may be differentiating. Due to the apparent absence of geographic barriers, the sympatric populations must be differentiating due to ecological differentiation [4], in this case, habitat preference. Indeed, the flexible genome provided evidence as L-population strains encoded flexible genes related to biofilm formation and the biosynthesis of MSHA for chitin adhesion. Thus, these closely-related strains are partitioning their microenvironments between particle-associated or free-living (migratory) populations. The acquisition of habitat-specific flexible genes can lead to ecological specialization, which further depresses recombination between the incipient populations. The question then becomes, can microscale environmental heterogeneity in microbial communities allow for the coexistence of closely-related organisms? Or put another way, are behavioral adaptations to particle-associated vs. free-living lifestyles strong enough to create boundaries to gene flow and structure population divergence? A follow-up study examined strains from both populations for various phenotypic measurements related to particle-association, including swimming speeds, cell sizes, flagellation, and chemotaxis [5]. Initially, strains from the L- population exhibited a differential ability to attach to particles (agarose, cellulose, alginate) and form biofilms (see below figure). However, S-populations strains (migratory, free-living) did not outcompete L-populations strains in swim speed nor chemotaxis in a steady resource gradient microfluidic chamber. Instead, the S-population strains, under time-varying conditions, were able to migrate to new nutrient supplies (really cool microfluidic videos here). Together, these closely-related strains appear to differentiate on fine-scale behavioral adaptations. 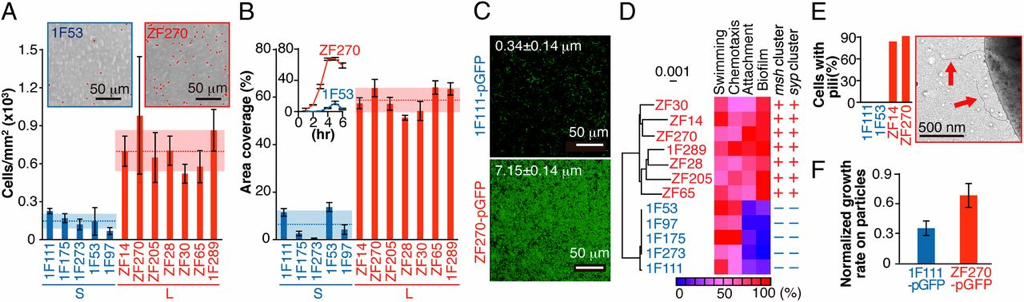 Phenotypic assays for S- and L-populations. A) Attachment to polystyrene. B) Biofilm formation and C) images. D) Correlation between S and L for phenotypic traits and genes. E) Number of pili F) Growth rate on alginate particles Papers1. Hunt DE, David LA, Gevers D, Preheim SP, Alm EJ, Polz MF. (2008). Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science 320: 1081–1085.
2. Datta MS, Sliwerska E, Gore J, Polz MF, Cordero OX. (2016). Microbial interactions lead to rapid micro-scale successions on model marine particles. Nature Communications 7: 11965. 3. Shapiro BJ, Friedman J, Cordero OX, Preheim SP, Timberlake SC, Szabó G, Polz MF, Alm EJ (2012). Population genomics of early events in the ecological differentiation of bacteria. Science 336: 48–51. 4. Cordero OX, Polz MF. (2014). Explaining microbial genomic diversity in light of evolutionary ecology. Nature Reviews Microbiology 12: 263–273. 5. Yawata Y, Cordero OX, Menolascina F, Hehemann JH, Polz MF, Stocker R. (2014). Competition–dispersal tradeoff ecologically differentiates recently speciated marine bacterioplankton populations. Proceedings to the National Academy of Sciences 111: 5622–5627. I am going to try and use this forum to give some thoughts on some recent publications I find interesting. I will likely list some of these papers in the blog, and hopefully find time to break down a few and highlight some of the bigger findings. Hopefully you all enjoy and definitely feel free to comment with suggestions!
|
AuthorSome thoughts on some (small) things Archives
May 2023
Categories |
 RSS Feed
RSS Feed