What is the biological question???I wanted to make a blog post about my recent microbiome workshop I led at Scripps Institution of Oceanography this week. The audience was mostly graduate students and faculty in my department whose research focuses on the discovery of natural products from marine organisms (see my previous post here about some cool research). I thought a lot of people might find this useful, especially earlier graduate students interested in microbiome work. So without further ado, here is a rundown of the workshop, including a nice dataset and some workflows I modified from the UCI Microbiome Initiative - special thanks to Julio and Claudia from the Whiteson and Martiny labs. Nowadays everyone is interested in how the microbiome affects [fill in your system here]. This had led to many studies including microbiome community analyses in their experimental design. Sometimes, the analysis does not coincide with the biological question (i.e., species level differentiation using 16S rRNA sampling). My point is to think carefully about what your biological question is and then design the correct sampling method to ensure the data can be interpreted and address your question. Also, there is a lot of limitations with each sequencing method (metagenomics v. amplicon-based) and understanding these limitations is paramount. I wrote another blogpost going over some of these topics. 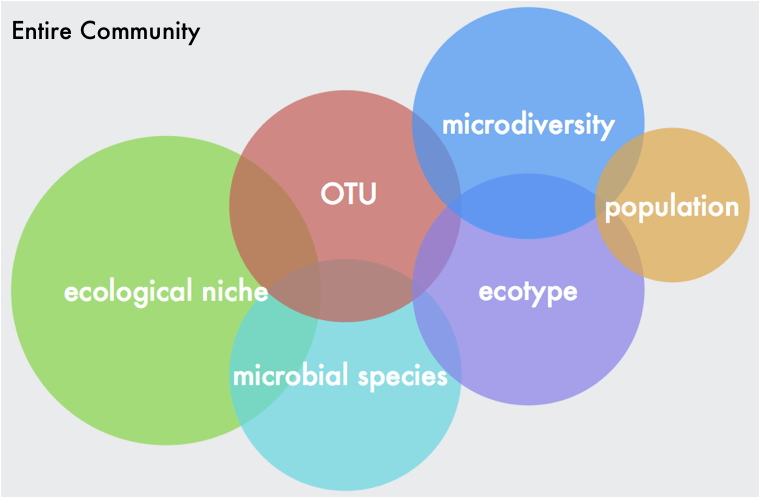 Briefly, it comes down to the degree of genetic resolution you want in your samples. For instance, the 16S rRNA gene has been instrumental in our understanding of microbial diversity. At the same time, it is a highly conserved marker gene. This means it cannot determine or resolve genetic (and definitely not functional) diversity of closely-related bacteria. “one limitation of the 16S rRNA gene is that it is rather conserved and hence is NOT reliable for taxonomic identifiers at the species level” -J. Cole et al. 2010.
Microbiome DataWhen you sequence a sample, you will get .fastq files or fasta files with quality scores at each base pair position (see powerpoint in github link below). These files will not only give you the DNA sequence information but also information on your sequencing run (e.g., how good is your data). This is where the first couple steps come into play. Whether you have 16S or metagenomic data, you will want to demultiplex your samples (assuming you ran a bunch of samples in a pooled library) and then denoise them. This means you will use those quality scores to trim your reads at a certain threshold. You will also want to trim the adapter sequences and/or primers off the beginning of each read. These steps can easily be done with tons of software - my personal favorite is BBMap but almost all of them will follow similar steps. 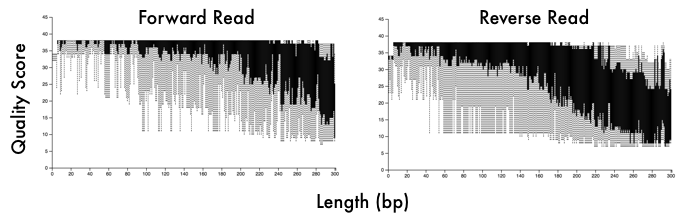 Typical visualization of QC (quality scores) of the fastq files. This data is 16S reads and we can see the adapters in the first 5 bp positions and then see a drastic decrease in quality scores at ~200bp for the forward reads and ~160bp for reverse reads 16S v Metagenomic OptionsMetagenomic Analysis So after you have your "cleaned up" data, what now? Well if you have metagenomic reads, you can perform assemblies and try to bin MAGs (metagenome assembled genomes - see post here). Or you can do a "read-based" approach to characterize the taxonomic diversity (like using MIDAS) or the functional diversity by searching for genes of interest. The latter, read-based approach, is nearly impossible to link function to taxonomy but can give you community-wide aggregated functional diversity. Metagenomic analyses can get really complicated so I will not go further into it here, but if interested here are some really useful links: Anvi'o by the Meren Lab Multi-Metagenome - R-based MAGs pipeline In general, you will need to follow these steps so learning how to navigate in a terminal is pretty essential. Here is some advice from a graduate student, Tyler Barnum at Berkeley. 1. Read assembly (MEGAHIT, metaSPAdes) 2. Read Mapping for coverage profiles (bowtie2, bwa, BBMap) 3. Binning 4. Bin Curation and Quality Check 16S rRNA amplicons This is where I wanted to focus this workshop. For one, I think most people will be analyzing this type of data. Furthermore, it is a lot more "user-friendly" to parse out this data. A ton of computational tools are available to analyze amplicon-based datasets, including QIIME2, DADA2, mothur, usearch, etc. each with tons of community support and tutorials. Post filtering and denoising, you will generally want to 1. Cluster sequences into operational taxonomic units (OTUs) 2. Create an OTU table with abundances of taxa across samples 3. Interpret data Most of the tools available have step-by-step instructions on how to navigate through data processing. These steps are pretty straight-forward and I included an example run in the github link below. One of the major decisions you will need to make during this process is whether to use traditionally-defined OTUs (97% OTUs) or use the growing application of single nucleotide variants (ESVs or ASVs or 100% OTUs). Either way, remember that the 16S has limited resolution and whatever clustering threshold you use cannot give you species level resolution nor intra-species patterns or processes. Don't make this mistake! In any case, your clustering threshold has a lot of debate and back-and-forth in the community so I will refrain from going into this as I am not typically analyzing 16S rRNA data. However, I definitely recommend you reading some information on this and educate yourself on the literature and the reasoning behind each argument. Workshop MaterialsAlright - that is enough background information. Here is the link to the workshop materials. I wanted to use R to analyze data as I think walking through this yourself gives a better understanding of what is happening. R has several advantages including reproducibility and many packages created specifically for microbiome analysis (e.g., phyloseq).
Github link: https://github.com/alex-b-chase/16S-workshop
0 Comments
Leave a Reply. |
AuthorSome thoughts on some (small) things Archives
May 2023
Categories |
 RSS Feed
RSS Feed