|
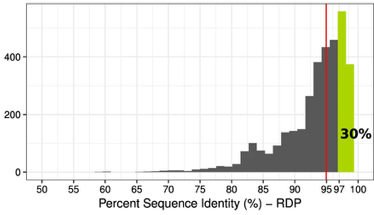
Recently, a lot of conversation has been circulating regarding what constitutes a microbe. How do we know we captured the diversity in a system and have a representative sample or sequence? Are abundant, relevant members of a community culturable? If not, how can we be sure that the computational output is representative? Especially with the increased use of culture-independent methods in microbiome research, we are relying heavily on the molecular and computational methods to assess this vast diversity of microorganisms. For this post, I will focus less on ideas of microdiversity (although this will come briefly later) within 16S defined OTU/ESVs. Instead, I want to highlight some of the more interesting conversations happening in microbial ecology regarding 1) culturability and 2) metagenome-assembled genomes or MAGs. This post largely spawned from the amazing dialogue being generated over Twitter over these two issues, and I just wanted to give my opinion on the topics. I will refrain from "taking sides" but focus on the arguments and the bigger picture implications for microbiome research. High proportions of bacteria are culturable across major biomesLet's start here because I think this is a SUPER important topic and obviously will relate to the second conversation below. For decades, microbiologists have leaned heavily on a major crutch: paradigm that only 1% of microorganisms are culturable A recent communication by Adam Martiny in ISME [1] challenged this paradigm in microbiology by arguing that, based on 16S rRNA similarity of bacteria, abundant members (35-52% of sequences and taxa, respectively) across major biomes have a representative member in a cultured isolate. At first, I was delighted to see this communication published. Just weeks before this, I saw multiple seminars and job talks starting the Introduction with highlighting this paradigm. Inevitably, it came across, that "well we can't culture bacteria, so why even try?". Subsequently, the talks would go over culture-independent approaches to navigate this "massive hurdle" in microbiology. But this is a major disservice to all the hardwork done to culture bacteria. Everything, in theory, is culturable - it's a matter of figuring out how to do it. The question shouldn't be is X bacterium culturable? it should be how do we culture it? This ideology of yet-to-be cultured microbes is definitely painstaking work, but it is possible (everyone please check out the incredible work of Yoichi Kamagata to characterize anaerobic bacteria and their syntrophic relationships - some culturing issues were resolved just by switching the preparation of the media! [2]).
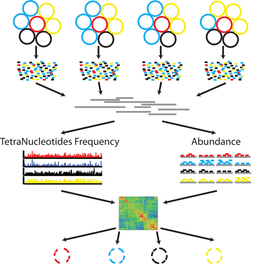
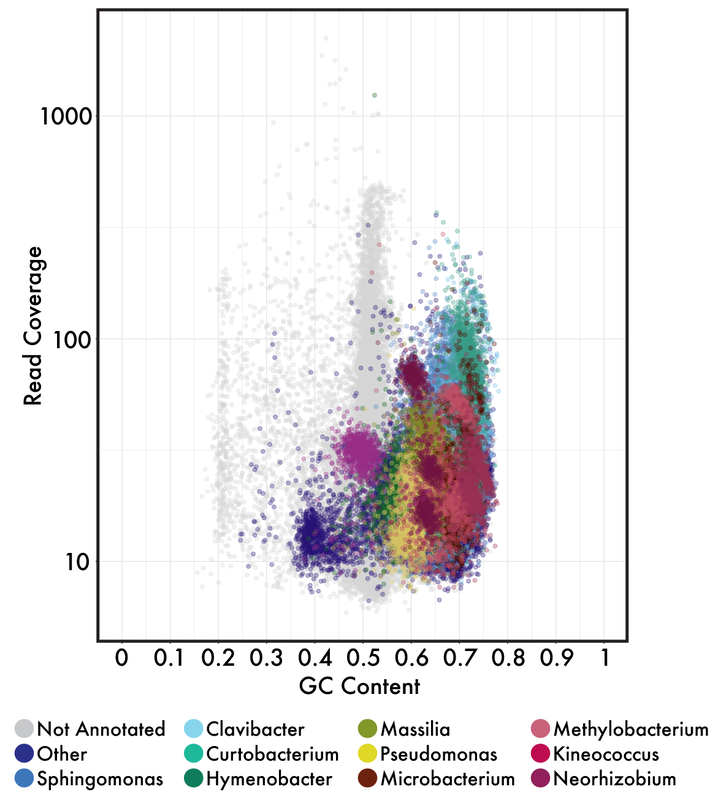
Both of these papers make great points and address topics that every microbiologists should be thinking about. As I have said before, many studies infer far too much from descriptive datasets of 16S rRNA community analyses. It is almost impossible to relate this data or the observed taxa to ecological function. I have spent a lot of time laying out some of these arguments, but basically comes down to the 16S rRNA gene provides a coarse depiction of the microbial community. There are millions of years of divergence and adaptation masked by the use of conserved marker genes. These are some of the similar points the Steen et al. paper lay out to argue against the idea that most organisms have been cultured. However, I do feel like this resolution, specifically with 16S rRNA studies, it is relevant to compare against databases. This is exactly what Martiny's response [4] details that the most common interpretation using a defined taxonomic unit (OTU) implies a lot of abundant members have cultured representatives in databases, certainly superseding the 1% "rule". In any case, how you define whether a taxa is cultured or what is the real number so far really depends on interpretation. And based on this interpretation, researchers need to apply appropriate methods. If you are concerned with fine-scale diversity, do not use 16S rRNA surveys. If you want to target a specific group, design taxa-specific primers. If you want to infer function and avoid PCR and molecular biases, probably switch to metagenomes. And finally, if you wish to link genotype to phenotype, figure out a way to culture and target the abundant members of your system. Physiological data can provide a lot and was the basis for most of my work to target isolates. Artificial construction of metagenome assembled genomes (MAGs)???I think this is a nice segue into the next topic. Based on the vastly different ways to interpret culturability of a microbe, I think it is super interesting that MAGs aren't more of a topic of discussion. For those who are unfamiliar, MAGs are extracted from metagenomic data using computational approaches to assemble "community" data, bin assembled contigs into similar sequences based on genomic signatures (e.g., %GC, tetranucleotide frequencies), and assess contamination to create MAGs of otherwise yet-to-be-cultured ;) bacteria or archaea (schematic below).
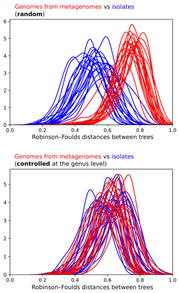
As many members were quick to point out, cultured isolates exist for the novel lineages where high quality MAGs (i.e., complete genomes) match pretty nicely. As an ultra bonus, one of the "artificial" lineages detailed in the preprint recently was isolated and described in another preprint documenting a 12 year(!) isolation effort for Asgard Archaea - this is why science is so awesome! Further, a really talented grad student in the Banfield Lab, Alex Crits-Christoph, re-analyzed this data to address the main support against MAGs in the preprint: The effect observed is due to differences in the phylogenetic distribution of genomes from metagenomes vs isolate genomes.
I personally think the authors vastly overstate their conclusions but I do think they bring up a few good points, especially if we start to bring in the ideas from the previous section. MAGs are useful because we can essentially sequence the entire microbial community and "pull out" genomes without isolating strains. This was incredibly useful in simple microbial communities where low genetic diversity allowed for the relatively easy binning of MAGs. However, I do start to question how much of MAGs, especially in complex, genetically-diverse systems, are a representation of a mosaic of closely-related strains and/or lineages.
Papers:1. Martiny AC. (2019). High proportions of bacteria are culturable across major biomes. ISME 13: 2125-2128.
2. Kato S, Yamagishi A, Daimon S, Kawasaki K, Tamaki H, Kitagawa W, Abe A, Tanaka M, Sone T, Asano K, Kamagata Y. (2018). Isolation of Previously Uncultured Slow-Growing Bacteria by Using a Simple Modification in the Preparation of Agar Media. Applied Environmental Microbiology 84: e00807–18. 3. Steen AD, Crits-Christoph A, Carini P, DeAngelis KM, Fierer N, Lloyd KG, Thrash JC. (2019). High proportions of bacteria and archaea across most biomes remain uncultured. ISME. 4. Martiny AC. (2019). The '1% culturability paradigm' needs to be carefully defined. ISME. 5. Garg SG, Kapust N, Lin W, Tria FDK, Nelson-Sathi S, Gould SB, Fan L, Zhu R, Zhang C, Martin WF. (2019). Anomalous phylogenetic behavior of ribosomal proteins in metagenome assembled genomes. bioRxiv. 6. Chase AB, Karaoz U, Brodie EL, Gomez-Lunar Z, Martiny AC, Martiny JBHM. (2017). Microdiversity of an abundant terrestrial bacterium encompasses extensive variation in ecologically relevant traits. mBio 8: e01809-17.
1 Comment
Alex
9/27/2019 12:41:06 pm
I am posting a useful comment from Twitter regarding this post from Alex Crits-Christoph ( Twitter @acritschristoph ): Leave a Reply. |
AuthorSome thoughts on some (small) things Archives
May 2023
Categories |
 RSS Feed
RSS Feed